How to use TCGA data to analyze intestinal flora
- A Persistent Crisis: The Looming Specter of Drug Shortages in United States
- Rabies: The fatality rate nearly 100% once symptoms appear
- Human Brain Continues to Grow: Study Shows Increase in Size and Complexity
- CRISPR Genome Editing: From Molecular Principles to Therapeutic Applications
- Metformin Helps Immune System Better Recognize Cancer Cells
- Highlights of Prostate Cancer Research at the 2024 EAU Congress
How to use TCGA data to analyze intestinal flora
How to use TCGA data to analyze intestinal flora. We know that the TCGA database includes the next-generation sequencing data of many patients. About next-generation sequencing. As we mentioned before, the data of second-generation sequencing is actually a part of the nucleotide sequence of all cells. For this part of the sequence, just look at the reference genome of what background species we take, and then we can compare what we get. Therefore, if the microbial flora genome is used, it is possible to obtain relevant data on the flora. So there is this article. So let’s briefly introduce this article first.

This article mainly uses Pathseq’s algorithm to evaluate the microbial flora based on the whole genome sequence (WGS) and whole exon sequence (WXS) data in gastrointestinal tumors of TCGA. However, there is always a bias based on human sequencing data, so most of the previous article is to remove these biases. After detailed operations, data on TCGA intestinal flora (The Cancer Microbiome Atlas, TCMA) was obtained.

Cluster analysis
After a series of algorithmic operations, the author obtained the microbial flora of each sample of the intestinal tract. Therefore, a simple analysis of these data is carried out. First, the author uses the SparCC algorithm to perform a cluster analysis of intestinal tumor CRC (COAD/READ). Finally, it was found that CRC can be divided into two clusters of Clostridium and Bacteroides.

Based on the classification of the two clusters, the influence of different flora on other omics RNA-seq, miRNA-seq, methylation, and RPPA was analyzed. Look for the possible mechanism of action of these flora.
Differential flora analysis and prognostic correlation analysis
Because CRC contains cancer and normal tissues. Therefore, the author also adopted a difference analysis to observe which flora is related to the occurrence of cancer. At the same time, because it also contains prognostic information, a prognostic analysis is also carried out to see which flora is related to the prognosis.
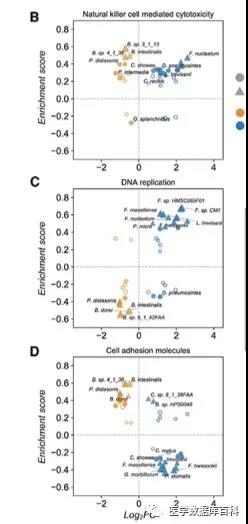
Further authors conducted an enrichment analysis on the differential flora. To learn more about which pathways these flora are related to. After analysis, it is found that the microorganisms in CRC are mainly related to the host immune response, inflammatory cancer pathways and cell-cell adhesion pathways.
TCMA database
The above is the main content of this article, of course, if it is, it can only explain what this article does. It may be of no use if we want to dig further. But after doing these analyses, this article also established a simple TCMA database (https://tcma.pratt.duke.edu/). It stores the results of all the author’s digestive tract tumor tissues after filtering analysis.
This database is mainly a place to download data. There is not much other analysis.

The gut microbial data downloaded from this database, plus other omics data downloaded from other places, are cross-analyzed through multiple omics. Still digging out something. So if you are studying gastrointestinal tumors. Can be used to analyze Kazakhstan.
(sourceinternet, reference only)
Disclaimer of medicaltrend.org