Pharmacological effects of CAR-T cells
- Mifepristone: A Safe and Effective Abortion Option Amidst Controversy
- Asbestos Detected in Buildings Damaged in Ukraine: Analyzed by Japanese Company
- New Ocrevus Subcutaneous Injection Therapy Shows Promising Results in Multiple Sclerosis Treatmen
- Dutch Man Infected with COVID-19 for 613 Days Dies: Accumulating Over 50 Virus Mutations
- Engineered Soybeans with Pig Protein: A Promising Alternative or Pandora’s Dish?
- Severe Fever with Thrombocytopenia Syndrome (SFTS): A Tick-Borne Threat with High Mortality
Pharmacological effects of CAR-T cells
Pharmacological effects of CAR-T cells. Compared with most drugs, CAR-T cell products are very complex, because each cell product is composed of a heterogeneous mixture of millions of cells.
Considering the ability of T cells to actively migrate and replicate in the patient’s body, the biodistribution and kinetics of CAR-T cells after administration are unique. CAR-T cell therapy also has multiple mechanisms of action, contributing to its anti-tumor activity and toxicity. This review summarizes the unique pharmacological effects of CAR-T cells, focusing on CAR-T cells that target CD19 and B cell maturation antigen (BCMA).
1 Introduction
Due to the development of molecular biology at the end of the 20th century, genetically engineered cell therapy emerged as a new type of treatment for cancer, genetic diseases, infectious diseases and autoimmune diseases. Among these therapies, chimeric antigen receptor (CAR) modified T cell (CAR-T cell) therapy is the most advanced method. The US Food and Drug Administration (FDA) approved two CAR-T cells in 2017 Therapy, this is the first regulatory approval of genetically engineered cell therapy.
CAR-T cells are very different from traditional drugs (such as small molecules or biological agents). Unlike the latter drug (which can be well defined chemically), cell-based therapies are extremely complex. Each cell in the product is composed of thousands of proteins. The abundance of these proteins not only varies from cell to cell in the product, but also varies with the cell’s response to its microenvironment. This complexity challenges our ability to define the purity and potency of cell products. The self-replicating nature of cell-based therapies is critical to their ability to produce long-term disease control, which also gives them unique pharmacological properties in drugs. This review describes the pharmacological effects of CAR-T cells and highlights some of the major gaps in our understanding of this rapidly developing field.
2. Overview of CAR-T cell therapy
CAR-T cell therapy uses genetically modified T cells to attack the patient’s tumor cells. They are based on the concept that engineered receptors composed of antibody-derived antigen binding domains and important T cell receptor signaling domains can guide T cells to react with antigen-expressing cells (2). Although the constructs of these engineered receptors (i.e. CARs) differ between different CAR-T cell therapies, CARs most commonly have single-chain variable fragments (scFv) as their extracellular antigen-binding domains. The intracellular signal transduction domain usually includes the T cell receptor CD3ζ domain and the costimulatory domain of CD28 or 4-1BB (as shown in Figure 1a).
After binding to specific antigens on tumor cells, the CAR signal domain will activate CAR-T cell proliferation, secrete cytokines or kill target tumor cells (3). The manufacture of most CAR-T cell products, especially those that have been approved by regulatory authorities, rely on the patient’s autologous T cells to avoid rejection of CAR-T cells or transplantation due to major and minor histocompatibility differences Material versus host disease. In a typical manufacturing process, the patient’s T cells are obtained by leukocyte separation, genetically modified in vitro to express CAR, and then amplified in cell culture, and finally transferred back to the patient to fight tumors (4 ) (Figure 1b).
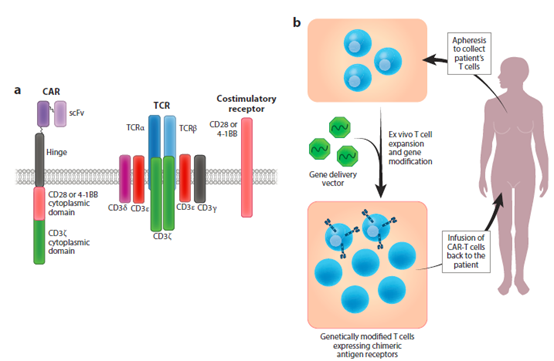
Figure 1 CAR design and its application in adoptive T cell immunotherapy. (A) Typical CAR design compared with natural TCR complex and costimulation. (B) Using genetic modification to express CA variable fragments; TCR, T cell receptor.
The most commonly used gene delivery vectors are γ-retroviral vectors or lentiviral vectors (see Table 1). Both types of viral vectors can carry out CAR-based R T cells in vitro for autologous T cell immunotherapy. Overview of general methods . Abbreviations: CAR, chimeric antigen receptor; scFv, single chain variable fragment; TCR, T cell receptor.
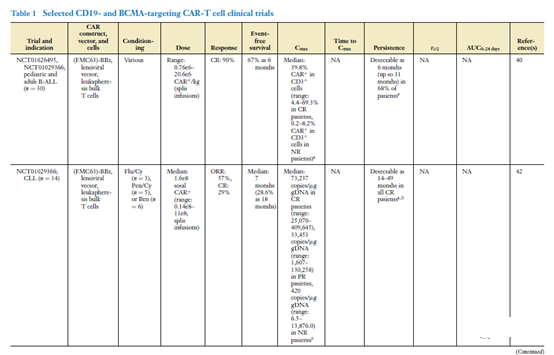
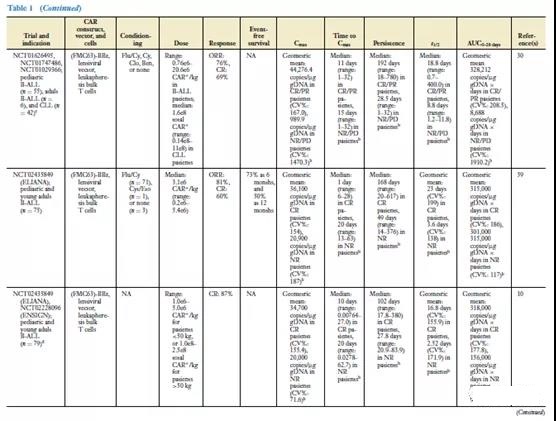
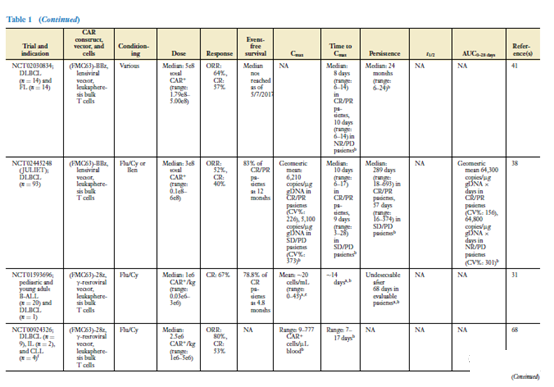
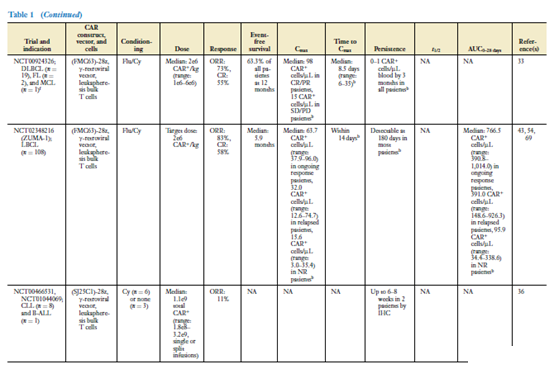
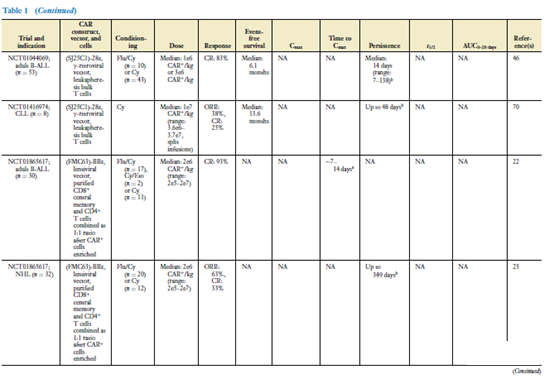
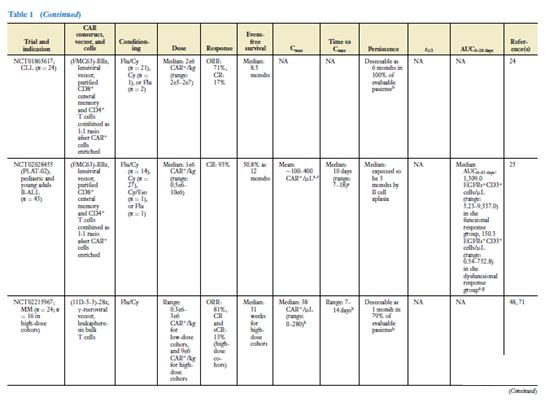
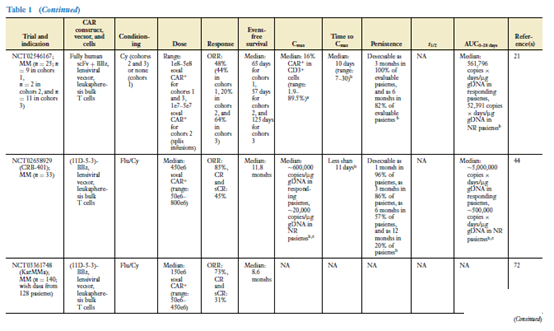
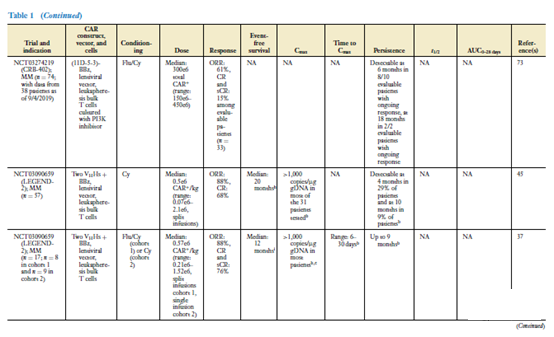
The most commonly used gene delivery vectors are γ-retroviral vectors or lentiviral vectors (see Table 1). Both types of viral vectors can effectively deliver CAR genes to stimulated human T cells in vitro. The main safety hazards of using viral vectors are the occurrence of viruses with replication capabilities and carcinogenesis due to the random insertion of genes into the genome of the vector. However, modern retroviral and lentiviral vector designs use split genomes for vector packaging, and integrate self-inactivating deletions in the viral genome, thereby reducing the acceptance of retrovirus or lentiviral vectors (5) in γ The possibility of replicating viruses in patients with T cell products modified by the receptor gene. In addition, there has been no report of carrier-mediated tumorigenesis in human T cells so far.
Several non-viral gene delivery methods for CAR-T cell manufacturing are also being studied, including Sleeping Beauty transposon system (6), piggyBac transposon system (7, 8) and messenger RNA (mRNA)-based systems ( 9).
Two analysis reports on CAR-T cells produced by lentiviral vectors show that the transduction efficiency has nothing to do with the in vivo kinetics of CAR-T cells (10, 11). Reference 12 provides a more comprehensive review of gene delivery vectors used in CAR-T cells. Reference 4 summarizes the production process and variation of other CAR-T cells.
The target antigen of CAR-T cells depends on the condition of the tumor. CD19 is an antigen widely expressed by cells of the B cell lineage. Another CAR target that has been extensively studied is the B cell maturation antigen (BCMA). BCMA exists on plasma cells, and CAR-T cell therapy targeting BCMA is actively studying R/R multiple myeloma (MM). In a number of clinical trials in the United States and China, CAR-T cells targeting BCMA have shown encouraging results (see Table 1).
Define the characteristics of living medicines
In contrast to traditional medicines defined by chemical composition (which can be used to evaluate the purity of products and predict their efficacy), CAR-T cells are composed of a variety of heterogeneous cells, which are derived from patients and can occur during the manufacturing process. Big change.
T cells are divided into two main subsets. CD8+ T cells mediate direct cytotoxic activity against target tumor cells through a variety of mechanisms, including the release of perforin and granzyme B and death receptor signaling, which trigger caspase activation in target cells and lead to apoptotic cell death (figure 2).
CD4+ T cells can assist this immune response by supporting CD8+ T cell proliferation and effector function cytokine production and direct cytotoxic activity (15, 16). These T cell subsets start from immature cells and gradually differentiate into a variety of memory or effector cells. Naive and memory T cells have the highest proliferation ability after stimulation, and differentiated effector cells coordinate the elimination of targeted tumor cells through dozens of different differentiation states in phenotype and function.
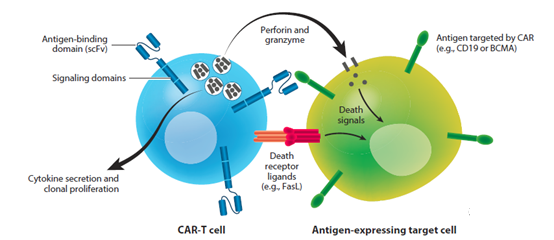
Figure 2 The mechanism of action of CAR-T cells. CAR most commonly consists of the extracellular antigen binding domain of scFv and the intracellular signaling domain, the latter usually including the TCRCD3ζ domain and the costimulatory domain of CD28 or 4-1BB. After binding to specific antigens on tumor cells, the CAR signal domain will activate CAR-T cell proliferation, secrete cytokines or kill target tumor cells. Abbreviations: BCMA, B cell maturation antigen; CAR, chimeric antigen receptor; scFv, single chain variable fragment; TCR, T cell receptor.
According to the Code of Federal Regulations, Title 21, Section 600.3, “Purity refers to the relative freedom from the influence of irrelevant substances in the finished product, whether harmful to the recipient or harmful to the product”. It is very difficult to define the purity of CAR-T cell products. In addition, because CAR-T cells are made from the autologous T cells of each patient, the starting materials vary greatly, which hinders the batch-to-batch consistency of the final product.
In addition to the heterogeneity that we can assess, there are many levels of change that cannot be measured. In the manufacturing process, random differences will inevitably be introduced into CAR-T cell products through gene delivery vectors, because most current methods (for example, γ-retroviral vectors, lentiviral vectors or non-viral transposon systems) Will result in the integration of genes at random locations within the genome.
Although carrier-mediated carcinogenesis in T cells has not been reported so far, the role of random gene insertion is still an uncontrollable factor that may affect the performance of CAR-T cells in patients. For example, it has been reported that random insertion events mediated by lentiviral vectors disrupted the methylcytosine dioxygenase TET2 gene in CAR-T cells, thereby epigenetically altering CAR-T cells and their progeny as well as the single The ability of cell clones to proliferate has been identified as the cause of patient remission (18). Although this patient benefits from the randomness caused by the gene delivery vector for CAR-T cells, it cannot be ruled out that mutations mediated by this vector may also have a negative impact on the efficacy of CAR-T cells.
In addition to purity, it is also important to predict product efficacy from a regulatory perspective and from the perspective of providing patients with the expected effective product. Many functional assays have been explored as potential potency assays for CAR-T cell products. The results of tisagenlecleucel’s report show that there is almost no correlation between the in vivo efficacy of CAR-T cells and their in vitro functions, which is determined by the production of interferon-γ (IFN-γ) that responds to tumor cells expressing the antigen (19) .
This is not surprising, because the production of this cytokine is not equivalent to cytotoxicity. In addition, much of the work to kill cancer cells after adoptive transfer of CAR-T cells is performed by the progeny of the injected cells rather than the injected cells themselves. Therefore, the in vivo efficacy of CAR-T cells may depend on their replication ability, which may depend on the memory differentiation subpopulation of CAR-T cells with high replication ability but less cytotoxicity or effector functions. Relevant studies conducted with tisagenlecleucel have proved this, showing that one of the best factors related to the efficacy of CAR-T cells is the CD27+ CD45RO-CD8+ T cells (a memory-like T cell) in the apheresis blood separation product used for CAR-T. Proportion of cells produced by cell subpopulations (20, 21).
Efforts are being made to incorporate cell selection into the CAR-T cell production plan, aiming to formulate CAR-T cell products with clearer ingredients and potency. In one method, purified CD8+ central memory T cells and CD4+ T cells were modified to express CAR, then CAR+ cells were enriched, and then the manufactured CD8+ and CD4+ CAR-T cells were combined according to a determined 1:1 ratio. Use a single product before injecting into the patient (22-25). This agreement is based on the results of preclinical studies, which show that CAR-T cells that target CD19 produced by selected central memory T cells or naive T cells are better than CAR-T produced by effector memory T cells when cleared.
The cells are more effective against CD19+ leukemia cells in a mouse model, and the combination of CD8+ and CD4+ CAR-T cells synergistically exerts an anti-tumor effect (26). However, this CD19-targeted CAR-T cell preparation has not yet shown higher clinical efficacy consistency in trials. In addition, the selection procedure for CD8+ memory T cells is not feasible in all patients (22). There is still a lot of work to be done to develop products with a clearer cell composition, which is likely to depend on a better understanding of the relationship between T cell biology and the quality and efficacy of CAR-T cell products.
Pharmacokinetics of CAR-T cells
CAR-T cells have strong proliferation ability after encountering target cells expressing antigens, and some CAR-T cells as memory cells may be able to last for many years in the body. These characteristics make the pharmacokinetics of CAR-T cells completely different from traditional drugs. The study of tisagenlecleucel and axiabtagene ciloleucel showed that the early kinetics after cell administration was similar. Usually, CAR-T cells will expand in large quantities after infusion, and their concentration in peripheral blood will reach a peak within 7 to 14 days after infusion. Then the concentration tends to decrease in two general stages. In the next few months after tumor clearance, an acute systole occurs, with a variable rate of decay. Then, in some patients, CAR-T cells showed detectable long-term persistence of CAR-T cells within a few years after the initial infusion (Figure 3; Table 1).
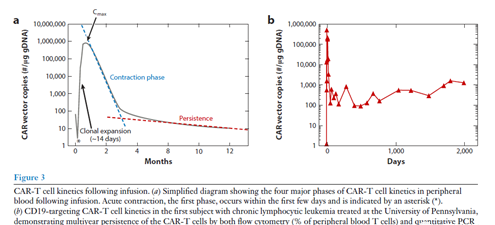
Several factors affect CAR-T cell dynamics in vivo. The first factor is inherent in the CAR structure that armed T cells. The costimulatory domain of CAR (usually from CD28 or 4-1BB) is essential for initiating and maintaining clonal T cell proliferation. However, the optimal costimulatory domain is not yet known and may depend on the condition of the tumor. The CD28 costimulatory domain stimulates CAR-T cell proliferation and enhances effector functions, especially cytokine production (27).
The 4-1BB costimulatory domain seems to promote the survival and persistence of CAR-T cells, especially in vivo (28). However, due to the various manufacturing protocols for CAR-T cell products, it is impossible to draw conclusions about which domain is superior using different CAR constructs to evaluate data in clinical trials. A study attempted to compare the engraftment kinetics between CD28 and 4-1BB co-stimulated CAR-T cells, where the two types of CAR-T cells were mixed in a 1:1 ratio before infusion into the patient, and Their implantation is simultaneously monitored by quantitative polymerase chain reaction (PCR).
This study did not find that in the two types of CAR-T cells implanted, the time to reach the expansion peak or the expansion amplitude determined by the area under the curve from day 0 to day 28 was statistically There is no significant difference (29). Nevertheless, this result may be due to the small cohort of the study. This study also did not evaluate long-term transplantation, because the difference in the persistence of CAR-T cells may be more pronounced. CAR-T cells targeting alternative antigens seem to show similar engraftment kinetics, as revealed by the similar in vivo kinetics of CAR-T cells targeting CD19 and BCMA produced by the same protocol.
The only difference is the antigen Binding domain (21, 30). Since many CAR constructs are composed of murine scFv as the antigen-binding domain, there are concerns about the potential immune rejection of CAR-T cells in patients. Pre-existing or induced anti-mouse purine antibodies have been observed, but they do not seem to affect the persistence or efficacy of CAR-T cells (10, 11, 31). However, the endogenous T cell response to murine scFv may hinder some implantation and efficacy of patients receiving the second CAR-T cell therapy (22, 23).
The difference in the quality of T cell subsets obtained from patients is the second important factor, which may affect the kinetics and long-term durability of CAR-T cell implantation. Some studies in the preclinical environment have shown that after adoptive transfer, naive, stem cell memory and central memory T cells show the highest persistence, and may be the best T cell subpopulation of CAR-T cells (32). A retrospective analysis of CLL subjects treated with CAR-T cell therapy with CD19 targeting and 4-1BB co-stimulation showed that the higher frequency of CD27+ CD45RO-CD8+ T cells in apheresis blood consumables was used to make CAR enhancement The expansion and persistence of CAR-T cells related to T cells and good clinical effects (20). A similar association was also observed in another study using CAR-T cells targeting BCMA to treat MM patients (21).
In CLL patients and MM patients, a higher CD4:CD8 ratio of T cells in apheresis products is also associated with greater CAR-T cell expansion (20, 21). No such correlation with CD4:CD8 ratio was found in the final CART cell products (10, 11, 20, 21). A study of CAR-T cells co-stimulated by CD28 showed that in patients with the following diseases, the percentage of central memory T cells in the CAR-T cell product (defined by CCR7+ CD45RA-) after infusion was compared with DLBCLCAR-T There is a correlation between cell peak expansion (33). In general, available data indicate that the in vivo kinetics of CAR-T cells may be affected by the composition of T cell subpopulations before and after manufacturing.
In addition to these CAR-T cell intrinsic factors, external factors can also affect CAR-T cell dynamics. According to reports, Tisagenlecleucel has a lower peak concentration in peripheral blood, but compared with B-ALL patients, DLBCL patients have longer persistence, which may be because CAR-T cells are transported between blood and tissues. Most target cells in DLBCL patients are located in the tissues, while most target cells in B-ALL patients are located in the blood.
This transport difference may lead to a decrease in blood concentration and a prolonged half-life of CAR-T cells (11). Tumor burden is another parameter that is often reported to be positively correlated with CAR T cell expansion (10, 22, 24, 30). In a study of CAR-T cells that target CD19 in B-ALL, the peak expansion of CAR-T cells is not related to overall tumor burden, but is related to CD19+ cells composed of tumor and non-tumor B cells in the bone marrow (25). These correlations can be explained by the presence of more antigen-expressing target cells, which leads to more frequent stimulation and proliferation of CAR-T cells.
However, in the study of CAR-T cells co-stimulated by CD28 in DLBCL, the relationship between tumor burden and antigen expression and CAR-T cell engraftment and dynamics has not been determined. This may be due to CAR-T cells targeting target tissues. Due to transportation. It may reduce the concentration of CAR-T cells in the blood sampled (11). Despite limited data, CAR-T cell dynamics does not appear to be affected by patient demographics (such as gender, age, race, and weight), tumor cytogenetics, disease stage, or previous treatment (10, 11).
Clinical factors that can be controlled to a certain extent will also change the kinetics of CAR-T cell implantation. Most notably, chemotherapy before injecting CAR-T cells will increase the peak value of T cell expansion. The value of patients receiving chemotherapy before T-cell immunotherapy was initially recognized in the study of tumor infiltrating lymphocytes (TILs) in melanoma. TIL treatment requires prior lymph node dissection to achieve the best clinical response (34).
The underlying mechanisms that enhance T cell expansion and engraftment after lymph node dissection chemotherapy are not fully understood, but they may be at least partly due to the increased availability of steady-state cytokines such as interleukin 7 (IL-7) and IL-15. Reduce endogenous host T cells’ competition for these limited cytokines (33, 35). The combination of cyclophosphamide and fludarabine represents the most commonly used chemotherapy regimen for lymphatic failure in CAR-T cell research.
Although cyclophosphamide alone can promote the implantation of CAR-T cells (36), the combination of fludarabine and cyclophosphamide has been shown to increase the growth of CD19-targeted CAR-T cells in adult B-ALL and NHL patients. Expansion and persistence and progression-free survival (22, 23). In addition, in a small study of CAR-T cells targeting BCMA in MM patients, compared with people without lymph node dissection, cyclophosphamide-mediated lymph node aspiration had neither CAR-T cell implantation nor No significant impact on CAR-T cell response, which indicates that although more consistent CAR-T cell expansion was observed in the lymph node dissection cohort, lymph nodes are not absolutely necessary for CAR-T cell implantation and persistence Dissection (21). Therefore, although preconditioning is generally considered to be an important part of T cell immunotherapy, further research is needed to better understand its full impact.
Finally, unlike most traditional drugs, the dose of infused CAR-T cells is not closely related to the in vivo kinetics of CAR-T cells in the patient (10, 11, 25, 33, 37-39). This may not be surprising, because CAR-T cells proliferate after the CAR binds to the relevant antigen and cause a logarithmic expansion of the number of CAR-T cells after infusion, which may offset any initial dose differences.
Pharmacokinetics of CAR-T cells
The clinical effects of CAR-T cells have been demonstrated in CAR-T cell products targeting CD19, and there are more and more data on CAR-T cells targeted by BCMA. Table 1 summarizes the data from most clinical trials targeting CAR-T cells against CD19- and BCMA. Disease histology is the main factor affecting the clinical response of CAR-T cells (35). The response rate of CAR-T cell products targeting CD19 to B-ALL patients is higher than that of DLBCL or CLL patients (38-42).
This may be due to differences in tumor location, characteristics of tumor cells, and tumor micro The quality of the patient’s T cells affected by the environment or the type of disease (30). The kinetics of clinical response also depends on the background of the disease. B-ALL and CLL patients treated with CAR-T cells targeting CD19 usually show the highest clinical response within the first month after infusion (30, 38, 43-45). However, delayed response may occur, sometimes even more than 1 month after the infusion (18).
The response does not seem to be related to patient characteristics such as race, gender, age, telomere length, or the number and type of previous treatments (20, 21, 38, 42, 45, 46). In addition, the target antigen density on tumor cells does not seem to affect the efficacy of CAR-T cells targeting CD19 or BCMA (21, 40, 42, 44, 45), and CD19-responsiveness has been observed in patients with DLBCL. The expression level is low or even undetectable (11, 38). The ability of low target antigen density to activate CAR-T cells is consistent with the observations of natural T cell receptors.
In this observation, a single peptide complexed with the major histocompatibility complex (MHC) can cause an activation signal (47) . This excellent sensitivity of CAR-T cells to antigen density can be contrasted with antibody-based therapies, which usually require a substantially higher target antigen density to achieve pharmacodynamic effects.
In clinical trials using various CAR-T cell products, the efficacy of CAR-T cells is positively correlated with the expansion and durability of CAR-T (10, 20, 21, 23, 24, 30, 31, 33, 38 , 42-44, 48), only one study showed that there is no clear relationship between the peak expansion of CAR-T cells and response in DLBCL patients (11, 38). Therefore, factors that enhance the expansion and persistence of CAR-T cells may contribute to the efficacy.
The correlation between the dose of CAR-T cells and the efficacy is not obvious (10, 11, 37-40, 42, 45, 46). Because CAR-T cells have the ability to proliferate, their expansion in the body is more important than the initial number injected into the patient. However, in several studies of CAR-T cells targeting BCMA in MM patients, although the sample size of these trials is relatively small, higher doses often lead to better clinical results (21, 44, 48).
As mentioned in Section 3, the retrospective analysis of the blood separation materials used to manufacture CAR-T cell products shows that the T cell subpopulation composition of the blood separation materials can predict the clinical outcome of CAR-T cell treatment with a higher frequency. The number of CD27+ CD45RO-CD8+ T cells in the raw materials for the manufacture of CAR-T cells is correlated with the improvement of clinical outcomes in patients with CLL and MM (20, 21). In contrast, retrospective analysis of CAR-T cells targeting CD19 in B-ALL showed that the higher frequency of LAG3 high/tumor necrosis factor-α (TNF-α)-low in apheresis blood wear materials CD8 + T cells and treatment failure (49).
In addition, it has been reported that the versatility of CAR-T cells before infusion (defined as the percentage of multifunctional cells multiplied by the median fluorescence intensity of cytokines secreted by those cells) is positively correlated with the clinical response of NHL patients (50). Whether these biomarkers can be used to predict the efficacy of the entire CAR-T cell product and disease, and how these phenotypes promote the improvement of efficacy, still needs further research.
Elucidated the mechanism of resistance to CAR-T cell therapy, that is, both primary resistance and secondary resistance will lead to relapse after treatment. At least two different mechanisms may cause primary resistance. First of all, treatment failure is attributed to the lack of CAR-T cell implantation and expansion, whether it is caused by the above-mentioned internal or external factors, this phenomenon is observed throughout CAR-T cell products and diseases, especially In CLL and MM (21, 42). Second, studies on B-ALL have shown that defects in death receptor signaling in leukemia cells may destroy the important role of tumors in CAR-T cell-mediated resistance, and these defects can destroy such as Fas-triggered apoptosis. And other death-inducing mechanisms. Although the release of perforin and granzyme B is considered to be the main cytotoxic mechanism used by CAR-T cells to eliminate tumor cells, they still have a killing effect (51).
Secondary drug resistance is most common in pediatric and young adult B-ALL patients, with a high frequency of clinical response (>80%) and a relatively high rate of disease recurrence (~40%) (35). In this case, relapse seems to be related to two main mechanisms. The first is to reduce the persistence of CAR-T cells, especially the early loss of CAR-T cells, which is related to the progression of antigen-positive tumors (6, 17, 20, 24, 30). The second is antigen loss, which accounts for about two-thirds of recurrent disease cases (10, 22, 40). There are many reasons for antigen loss. The main mechanism is mutation or deletion in the cd19 locus, which destroys the expression of the CD19 antigen or the antibody binding epitope (20). Compared with patients treated with CD28 costimulated CAR-T cells, the response of B-ALL patients treated with 4-1BB costimulated CAR-T cells was more durable (35). However, regardless of the persistence of CAR-T cells, the loss of antigens on tumor cells may lead to recurrence, which is more common in patients treated with CAR-T cells costimulated by 4-1BB. This may be due to their persistence. Improved (35).
Side effects
Cytokine release syndrome (CRS) and neurotoxicity are the most prominent adverse events associated with CAR-T cells targeting CD19 and BCMA (Supplementary Table 1). CRS is characterized by a surge of inflammatory cytokines (including IL-6, IFN-γ, TNF-α, IL-2, and IL-10), which are produced by activated lymphocytes and myeloid cells, and cause fever including fever , Symptoms including hypotension, hypoxia and organ dysfunction (52, 53). The onset of CRS may be rapid, and fever is one of the earliest symptoms.
In the registration trial of the abciximab gene iloleucel in DLBCL, the median time to onset of CRS was 2 days (range 1 to 12 days), and the median duration was 8 days. Similar kinetics were observed in DLBCL patients and pediatric and young adult patients with B-ALL. The severity of CRS ranges from mild fever to life-threatening, systemic inflammation associated with hypoxia, acute respiratory failure, and hypotension requiring intensive care. In most cases, CRS can be completely resolved without sequelae.
Many studies have used several severe CRS scoring systems with different criteria (52), which makes it difficult to directly compare the frequency of severe CRS in the entire clinical trial. However, as shown in Supplementary Table 1, the incidence of all grades of CRS is very high. In CAR-T cell trials for CD19 and BCMA, 50-100% of treated patients experienced CRS. Despite the challenges of comparing performance, severe CRS (grades 3-4) still occurs in 0-47% of patients. Fatal CRS is rare, but it can happen. Although no fatal events were observed in DLBCL (111 subjects) or pediatric and young adult ALL (75 subjects) in the Tisagan micronucleus registry trial, the axicarbabatan gene extracellular penicillin registry trial Two grade 5 CRS events (~2%) were reported in 101 subjects receiving treatment, one subject experienced phagocytic lymphocytic histiocytosis-like syndrome, and the other subject Experienced cardiac arrest (54). The American Society for Transplantation and Cell Therapy recently developed a consensus scoring system for CRS, which should help improve the comparison of CRS severity in all trials in the future (53).
CRS is highly correlated with serum IL-6 levels and responds to IL-6 blocking therapies such as tocilizumab. This drug and tisagenlecleucel have obtained regulatory approval for CRS management at the same time. Considering the potential severity of CRS, other methods have been explored to mitigate the risk of severe CRS. For example, in some trials, CAR-T cells were administered in divided doses over 3 days instead of a single full-dose infusion (Table 1; Supplementary Table 1), but this strategy lacks evidence to support its effectiveness. Another study used CAR constructs with low affinity for CD19 and reported that CAR-T cells with this CAR design did not induce severe CRS or neurotoxicity (57). However, the tumor burden of the patients participating in this study was relatively low, which confuses the comparison of this CAR design with other CAR designs. Several groups have attempted to identify biomarkers that can predict CRS, but this discovery is challenged by the rapid onset of CRS (21, 22, 24, 31, 40, 42, 44).
Neurotoxicity is another relatively frequent adverse reaction of CAR-T cells, which was not anticipated by preclinical studies. From mild irritation, headache and aphasia to reduced consciousness, tremor, epilepsy, and cerebral edema, neurotoxicity can manifest as a series of neurological symptoms (58, 59). Neurotoxicity tends to occur later than CRS (40), and most studies have shown that the median time to onset is 4 to 6 days (38, 43, 46, 58, 59). Most of the observed neurotoxicity is self-limiting and reversible, but fatal neurotoxicity has been reported (41).
The mechanism of neurotoxicity has not yet been fully revealed. Two studies (58, 59) have associated severe neurotoxicity with endothelial cell activation and blood-brain barrier dysfunction, which may allow inflammatory cytokines to enter the central nervous system (CNS). The presence of CAR-T cells in the central nervous system does not seem to be related to neurotoxicity (30, 40, 42, 59). However, the possibility that T cell activation occurs locally in the CNS via B cells or plasma cells in the CNS has not been completely ruled out. Like CRS, neurotoxicity is related to CAR-T cell expansion (23, 24, 30, 33, 46, 54) and tumor burden (22, 24, 30, 46), although evidence suggests that it is related to CAR-T cell dose Inconsistent (compare 10, 11, and 25 with 21-23). Although severe neurotoxicity is usually associated with concurrent CRS (39, 58, 59) and increased levels of IL-6 and IFN-γ (21-24, 33), neurotoxicity is less responsive to tocilizumab treatment (40, 58, 59) and it lacks clear intervention measures. Therefore, it is necessary to further study the mechanisms and treatments of neurotoxicity to improve the safety of CAR-T cells.
Finally, B-cell hypoplasia is an expected side effect of CD19-directed therapy, which occurs in most patients with persistent CAR-T cell implantation. In patients with B-ALL, it has been considered that the lack of B cells is a biomarker of the persistence of CAR-T cell function. In these patients, the early recovery of B cells is associated with antigen-positive recurrence (10, 40). Despite the lack of normal B cells, humoral immunity can be sustained due to the presence of long-lived plasma cells, which lack CD19 expression (60).
Sum up :
CAR-T cell-based therapies are novel therapies that are different from traditional drug therapies, and these therapies are rapidly becoming the standard of care for several B-cell malignancies. Although initially developed in the R/R disease environment, their position in the treatment paradigm may change, and the trial has evaluated tisagenlecleucel as a first-line high-risk B-ALL therapy.
There are still many questions surrounding the mechanisms underlying the unique toxicity observed with these therapies. It is very necessary to prevent these serious adverse effects. There is evidence that prophylactic tocilizumab may be able to reduce the severity of CRS without reducing anti-leukemia function (56, 65).
As our understanding of the mechanism increases, strategies using advanced gene editing and cell engineering methods may become feasible for eliminating key T cell-derived cytokines (such as IL-1 and GM-CSF), and the latter may be An important driving force for CRS (66, 67). From more than 400 different clinical trials exploring CAR-T cell treatment methods, it is also obvious that more of these therapies will be approved by the regulatory authorities, especially the CAR-T cell therapy for BCMA has shown excellent performance Clinical activity, despite facing persistent and possibly unique challenges to durability (Table 1).
(source:internet, reference only)
Disclaimer of medicaltrend.org