The diversity of coronavirus in a single host is underestimated
- Did Cloud Seeding Unleash a Deluge in Dubai?
- Scientists Identify Gut Bacteria and Metabolites that Lower Diabetes Risk
- OpenAI’s Model Matches Doctors in Assessing Eye Conditions
- UK: A Smoke-Free Generation by Banning Sales to Those Born After 2009
- Deadly Mutation: A New Monkeypox Variant Emerges in the DRC
- EPA Announces First-Ever Regulation for “Forever Chemicals” in Drinking Water
The diversity of coronavirus in a single host is underestimated
The diversity of coronavirus in a single host is underestimated.Researchers from the National Institute of Allergy and Infectious Diseases (NIAID) Vaccine Research Center published a research paper on PLoS Pathog on April 8.
The topic is High-throughput, single-copy sequencing reveals SARS-CoV-2 spike variants coincident with Mounting humoral immunity during acute COVID-19 (high-throughput, single-copy sequencing shows that the mutation of the new coronavirus spike protein occurs simultaneously with the increasing humoral immunity during the acute infection of new coronavirus pneumonia) (Reference 1).
The author’s conclusion is that the diversity of the new coronavirus in a single host is underestimated.
In the early days of the COVID-19 (First death in Canada after receiving AstraZeneca COVID-19 vaccine! pneumonia) pandemic, the mutation rate of the SARS-CoV-2 (First death in Canada after receiving AstraZeneca COVID-19 vaccine! virus) genome seemed to be very low, with an average of only 10 bases difference between the approximately 30,000 base genomes of any two isolates.
By the end of 2020, as the number of infected people increases, more variants will be isolated than previously seen.
Tracking the evolution of SARS-CoV-2 in infected individuals will help clarify the pathogenesis of COVID-19 and provide information for the use of antiviral interventions. A genomic-level mutation study showed that the diversity of new coronaviruses in a single host may be underestimated .
SARS-CoV-2 mutations will definitely appear in every infected person. These mutations may be affected by selective pressure, such as the pressure exerted by antibodies that bind to the spike protein. Commonly used sequencing technologies cannot detect different virus variants in a single sample.
In order to solve this limitation, the author of this paper has developed a high-throughput SGS (HT-SGS) strategy. SGS stands for single-genome amplification and sequencing. This method can extract a large number of single virus genomes. Deep sequencing of long reads on gene regions of surface proteins in.
The method is conceptually similar to the conventional SGS program, that is, a single molecule is amplified for Sanger sequencing in the case of limited dilution.
This method was applied to the control strain WA-1 of SARS-CoV-2 clinical isolates passaged in vitro and the upper respiratory tract samples of 7 COVID-19 study participants. The results show that during acute infection, the SARS-CoV-2 gene variant strain will appear under the immune pressure of the host. SARS-CoV-2 variants appear in every host and are affected by immune selective pressure.
This technique revealed that the clinical isolates of SARS-CoV-2 showed extensive genetic diversity after cell culture.
When this method is used to analyze the products of patient isolates (including SARS-CoV-2) passed 4 passages in Vero cells, a total of 18 unique types were detected in the scramble, ORF3, envelope, and M protein coding regions The combination of mutations constitutes 18 haplotypes, and there are aggregations of non-synonymous mutations in the cleavage site region of the NH2 terminal domain (NTD) of the spike protein (NTD) and furin.
More than half of the single-genome sequences differed from the reference sequence. Many changes can lead to amino acid substitutions, indicating that selective pressure plays a role in the adaptation process of cell culture.
Then, 7 patients were tested for virus variant strains within the host individual at 8-17 days after the onset of the disease. The diversity was low, and only a small number of haplotypes with the same sequence were detected.
These relatively homogeneous sequences may be the result of sampling during the early stages of infection when antibody pressure is limited. Testing the antibody response of the longitudinal sample showed increased and expanded antibody response to the spike protein.
Longitudinal analysis of an individual revealed 4 viral haplotypes, with 3 independent mutations in an NTD spike epitope targeted by autoantibodies (see figure below). Simultaneously with these mutations is a 6.2-fold increase in the ability of serum antibodies to bind to spurs and a transient increase in viral load.
The authors concluded that SARS-CoV-2 shows a rapid genetic adaptability that can be detected in the body when humoral immunity starts to work, which may lead to delayed virus clearance during an acute onset of the disease.
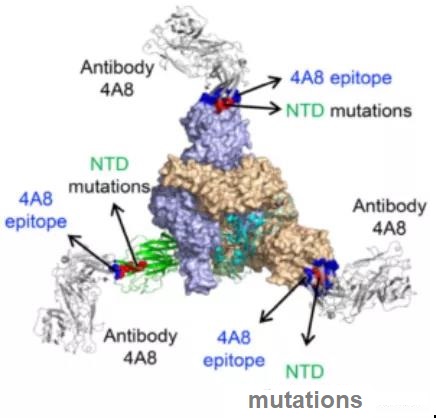
Figure: The interaction between the new coronavirus spike protein antigen and antibody. NTD mutation: NH2 terminal domain mutation; epitope: epitope (antigenic determinant); Antibody: antibody.
Studies on antibody responses and individual genome mutations have shown that there is a correlation between the appearance of antibodies to the N-terminal fragment of the spike protein, the amino acid changes that bind to these antibody regions, and the delay in virus clearance.
These results are consistent with the antibody pressure that causes SARS-CoV-2 to mutate during infection (as shown in the figure above). The variation of SARS-CoV-2 in a single host has not been described before, which may be due to the inability of previous sequencing methods to distinguish between variation and technical errors. It is also possible that high-quality data was obtained at an early stage of infection when virus titers are high and antibody pressure has not yet occurred.
With the increase in the ability of serum to bind to the spike protein, multiple SARS-CoV-2 variants can be detected with independent mutations on a single epitope, as well as a transient increase in viral load. These findings indicate that SARS-CoV-2 replication has resulted in sufficient viral genetic diversity to allow immune-mediated mutations to undergo natural selection during acute COVID-19 infection. Some important information can be derived from these findings.
They illustrate that the diversity of virus strains in a single host may lead to the emergence of antibody-resistant mutants. If the resulting mutants are more adaptable, they may spread more quickly in the population. In the early stages of infection, through previous immunization or antiviral therapy, before antibody selection occurs, virus propagation can be restricted to prevent the virus from escaping in the host.
This research method should be more applied to the study of COVID-19 patient samples to understand the potential variation of SARS-CoV-2 in the host individual, which will help determine the evolution of SARS-CoV-2 in the individual to COVID-19 The degree of influence on clinical outcome and antiviral drug sensitivity.
(source:internet, reference only)
Disclaimer of medicaltrend.org