Post-translational modification of cardiac sodium channel Nav1.5
- “Miracle Weight-loss Drug” Semaglutide Is Not Always Effective
- Study shows regular seafood consumption may increase risk of exposure to PFAS
- A Persistent Crisis: The Looming Specter of Drug Shortages in United States
- Rabies: The fatality rate nearly 100% once symptoms appear
- Human Brain Continues to Grow: Study Shows Increase in Size and Complexity
- CRISPR Genome Editing: From Molecular Principles to Therapeutic Applications
Research progress on post-translational modification of cardiac sodium channel Nav1.5
Research progress on post-translational modification of cardiac sodium channel Nav1.5. The cardiac sodium channel Nav1.5 is a key protein responsible for the formation of action potentials and impulse conduction in cardiomyocytes, and its functional status is jointly regulated by genetic factors and post-translational modifications.
Various post-translational modification methods including phosphorylation, glycosylation, acetylation, palmitoylation, ubiquitination modification, etc. can individually or cooperatively regulate the expression, positioning and channel dynamics of Nav1.5. The abnormality of Nav1.5 post-translational modification is also an important mechanism for triggering arrhythmia, which provides new ideas for the analysis and treatment of arrhythmia.
The cardiac voltage-gated sodium channel is composed of α subunit (namely Nav1.5) and β subunit, which are distributed in the interstitial disc, transverse tube and lateral membrane of cardiomyocytes, and are mainly involved in the formation of action potentials and impulse conduction.
Among them, the α subunit is coded by the SCN5A gene, and the four α subunits pass through the membrane six times to form a sodium ion shuttle channel, and the single β subunits across the membrane cooperatively regulate the opening of the channel. Mutations in the Nav1.5 gene can change the electrophysiological properties of sodium channels, causing long QT syndrome (LQTS), Brugada syndrome (brugada syndrome, BrS), atrial fibrillation, idiopathic ventricular fibrillation and sudden infant death syndrome (Sudden infant death syndrome, SIDS) and many other arrhythmias. In addition to gene regulation, Nav1.5 is also affected by post-translational modifications (PTM) such as phosphorylation, glycosylation, acetylation, and ubiquitination.
PTM promotes the functional maturation of proteins by covalently binding chemical small molecular groups on the amino acid side chains of proteins. In Nav1.5, PTM can regulate the number, location and function ofResearch progress on post-translational modificatio channel proteins through complex cellular and molecular mechanisms, thereby affecting the electrophysiological properties of cardiomyocytes. The schematic diagram of the PTM site of the cardiac voltage-gated sodium channel Nav1.5 is shown in Figure 1.
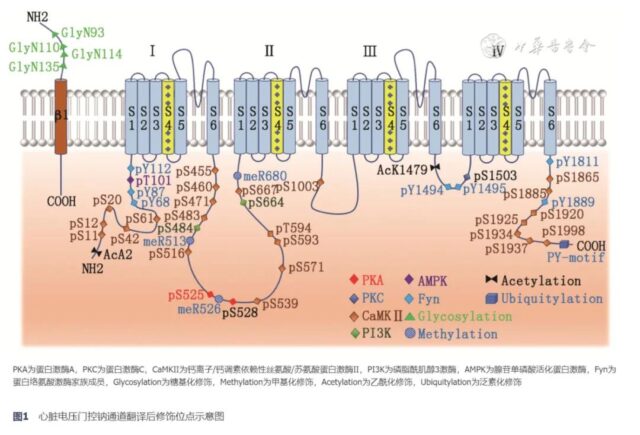
1. Phosphorylation modification
Phosphorylation modification is the most widely studied sodium channel PTM. A variety of protein kinases can introduce negatively charged phosphate groups into Nav1.5 through different ways. Among them, protein kinase A (PKA) and protein kinase C (Protein kinase C, PKC), calcium ion/calmodulin-dependent serine/threonine protein kinase II (Ca2+/calmodulin-dependent kinase II, CaMK II), phosphoinositide 3-kinase (PI3K) And adenosine monophosphate-activated protein kinase (5′ adenosine monophosphate-activated protein kinase, AMPK) is more common. Multiple phosphorylation modification pathways are not completely independent, and can modify the same amino acid epitope together to coordinately regulate the functional state of sodium channels.
1) PKA-mediated phosphorylation modification:
PKA is a common cyclic adenosine monophosphate (cAMP) dependent protein kinase. Under the connection of A-kinase anchor protein, it can convert adenosine 5′-triphosphate (ATP) into The γ-phosphate is transferred to the target protein to achieve phosphorylation modification [1]. PKA mediates the phosphorylation of Serine S525 and S528 in Nav1.5, which can promote the opening of sodium channels. At the same time, the phosphorylated serine residues increase the negative charge, which can cover the endoplasmic reticulum of Nav1.5.
The retention signal R533RR535 promotes its release from the endoplasmic reticulum and migration to the cell membrane, thereby increasing the number of sodium channels on the membrane surface [2]. In type 3 LQTS, the D1790G mutation in Nav1.5 can enhance the PKA-mediated phosphorylation modification, leading to the increase of late sodium current INaL, the prolongation of the action potential plateau and the QT interval. On the contrary, in BrS, the R526H mutation of the sodium channel can significantly inhibit the phosphorylation modification of S528, and down-regulate the number of sodium channels and sodium current density.
Studies have found that a frame-shift mutation in the coding gene NPPA of atrial natriuretic peptide precursor can also indirectly inhibit PKA-mediated phosphorylation modification and reduce sodium current [3]. However, the molecular mechanism of the above-mentioned mutations affecting phosphorylation modification and whether there are more PKA modification sites in Nav1.5 remains to be further clarified.
2) PKC-mediated phosphorylation modification:
PKC is a group of phospholipid-dependent calcium-activated serine/threonine kinases, regulated by circulating hormones such as angiotensin II and endothelin. Different PKC subtypes exert different biological effects:
(1) PKC-δ can phosphorylate the Serine S1503 site in Nav1.5, causing hyperpolarization of the sodium channel and reducing the opening frequency of the channel [4].
(2) PKC-α and PKC-ε can inhibit the forward transport of Nav1.5 from the endoplasmic reticulum to the cell membrane, reduce the number of sodium channels on the membrane surface, and reduce the peak current, but have no effect on the gating characteristics of sodium channels [5 ].
(3) The increase in calcium ions or reactive oxygen species in ventricular myocytes can activate calcium-sensitive PKC, delay sodium channel inactivation, and increase INaL [6]. Diseases related to abnormal PKC phosphorylation pathways are rare.
In BrS and SIDS, mutations in the gene encoding glycerol-3-phosphate dehydrogenase 1 like (GPD1L) can enhance the phosphorylation modification of PKC, leading to down-regulation of sodium channels and sodium current [ 7]. McCauley et al. [8] found in obesity-related atrial fibrillation patients that the up-regulation of PKC-α/δ expression can also inhibit the function of sodium channels, suggesting that blocking the phosphorylation modification pathway of PKC is expected to become the treatment approach for the above-mentioned arrhythmia.
3) CaMKⅡ-mediated phosphorylation modification:
CaMKⅡ is a serine/threonine protein kinase with multiple biological functions. CaMKⅡδc is activated under the control of calcium ions, which can phosphorylate the serine S516, 571, 593 and threonine T594 positions in Nav1.5, promote the shift of the steady-state inactivation curve to the hyperpolarization direction, and delay the rapid deactivation of sodium channels. Live, increase the peak sodium current and INaL [9, 10].
The increase of INaL can reduce the repolarization reserve, prolong the duration of the action potential, and trigger the early depolarization. The corresponding sodium overload can induce delayed depolarization, and through the sodium-calcium exchanger, increase the intracellular calcium ion level and further activate The biological function of CaMKⅡ. Burel et al. [11] found that CaMKⅡ and Nav1.5 are co-localized on the myocardial disc and T tube.
Overexpression of CaMKⅡδc can increase the risk of ventricular arrhythmia and heart failure. In AC3-I transgenic mice, inhibiting the function of CaMKⅡ can effectively improve the phosphorylation effect and electrophysiological changes [12]. In LQTS, the A572D and Q573E mutations in Nav1.5 will increase the level of phosphorylation near the S571 site of serine, mimicking the negative charge effect after phosphorylation of S571, leading to an increase in INaL and triggering the arrhythmia phenotype. Ranolazine can improve the electrophysiological effects caused by the above mutations by inhibiting INaL [13].
4) PI3K-mediated phosphorylation modification:
PI3K is a type of protein kinase with serine/threonine kinase activity and phosphatidylinositol kinase activity. It can be phosphorylated to modify protein kinase B (PKB) and 3 -Phosphoinositide-dependent protein kinase-1 (3-Phosphoinositide-dependent Protein Kinase-1, PDK1), indirectly regulates the function of Nav1.5. Serum and glucocorticoid-inducible kinase (SGK) acts as a downstream substrate of PDK1 and has serine/threonine kinase activity.
Through the PI3K-PDK1-SGK signal pathway, the S484 and S664 sites of Serine of Nav1.5 can be phosphorylated to facilitate sodium channel activation, increase sodium current, depolarize the inactivation curve, and reduce INaL [14]. In patients with diabetes and drug-induced LQTS, the prolongation of the action potential duration is closely related to the increase of INaL after the PI3K phosphorylation pathway is damaged [15].
Knockout of PI3K or direct inhibition of PKB can significantly increase INaL and trigger arrhythmia. Although both PKB and PDK1 are downstream substrates of PI3K, whether the above-mentioned kinases (PI3K, PKB, and PDK1) act synergistically or independently remains to be studied in depth.
5)AMPK-mediated phosphorylation modification:
AMPK is a key molecule in the regulation of bioenergy metabolism. Activated AMPK can delay the inactivation of Nav1.5, increase the continuous sodium current, and shift the activation curve to the hyperpolarization direction. Therefore, overexpression of AMPK can significantly extend the duration of action potentials and trigger early and later depolarization, which is also an important reason for the high risk of arrhythmia in patients with AMPK mutation-related pre-excitation syndrome [16].
In cardiac ischemia-reperfusion injury, activated AMPK can phosphorylate the threonine T101 site in Nav1.5, expose the adapting epitope of autophagy protein LC3, and induce the autophagic degradation of Nav1.5 by LC3, thereby reducing The number of sodium channels on the membrane surface, but whether it affects the occurrence of arrhythmia is still lacking in-depth research [17].
6) Fyn-mediated phosphorylation modification:
Fyn is a member of the non-receptor tyrosine kinase family, which can be phosphorylated to modify the N-terminal (Y68, Y87 and Y112), ICLⅢ-IV (Y1494, Y1495) of Nav1.5 And the tyrosine residues at the C-terminal (Y1811, Y1889) cause the depolarization shift of the steady-state inactivation curve, shorten the inactivation recovery time, and decrease the proportion of inactive channels, thereby enhancing the sodium current [18 , 19].
The above changes can be reversed by giving tyrosine kinase inhibitor (genistein/AG957/PP2) or tyrosine phosphatase PTPH1. In addition, Fyn can also increase the activity of PKA, indirectly exerting effects by affecting the downstream substrates targeted by PKA [20]. There are no reports of arrhythmia with abnormal Fyn pathway in clinical practice. Due to the low degree of tyrosine phosphorylation, limited by analytical methods, its role in sodium channels may be underestimated.
2. Glycosylation modification
Glycosylation modification refers to the process of transferring sugars to specific amino acid residues to form glycosidic bonds under the action of glycosyltransferase. The N-terminal asparagine 93, 110, 114, and 135 of the β1 subunit of the cardiac sodium channel bind to glycans under the control of sialic acid, which can promote the rapid inactivation of Nav1.5 and delay the resuscitation.
After knocking out the sialyltransferase gene ST3Gal4, due to the lack of sialic acid to regulate sodium channels, the open time and open rate of sodium channels increase, resulting in a shortened refractory period of cardiomyocytes and an increased susceptibility to ventricular arrhythmia [21, 22] .
The glycosylation modification of the B2 subunit is the key link for the efficient transport of Nav1.5 to the cell membrane. Studies have found that the non-glycosylated β2 subunits are mainly retained in the endoplasmic reticulum, and the rate at which they bypass the Golgi and reach the cell surface is about one third of the glycosylated modified subunits [23].
In addition, there are histological differences in the glycosylation modification of Nav1.5. Arakel et al. [24] found that the sodium channel glycosylation level of atrial tissue was significantly higher than that of the ventricle, suggesting that the difference in glycosylation modification may also be involved in the differentiation and maturation of the heart cavity.
3. methylation and acetylation modification
Methylation modification refers to the active methyl transfer process catalyzed by methyltransferase. Arginine R513, R526 and R680 in Nav1.5 are the main methylation modification sites. Overexpression of arginine methyltransferase can increase the level of methylation modification and increase the number of Nav1.5 on the cell surface. And sodium current intensity, induce BrS, type 3 LQTS and heart failure [25, 26]. Methylation modification and neighboring phosphorylation modification are mutually regulated. S516 phosphorylation can block R513 methylation, and R513 methylation can reduce S516 phosphorylation. Breaking the balance between the two can increase the risk of cardiac conduction system disease. [27].
Acetylation modification refers to a process in which the acetyl group of acetyl-CoA is reversibly transferred to an amino acid residue. After the lysine K1479 in Nav1.5 is deacetylated or mutated into a non-acetylated residue state, it can promote the migration of Nav1.5 in the endoplasmic reticulum to the cell membrane and increase the sodium current density [28]. On the contrary, when the mutation is in a continuous acetylation state, the sodium current can be significantly inhibited. In patients with conduction system diseases, the high acetylation of K1479 exists in Nav1.5, suggesting that this modification plays a role in the regulation of cardiac electrical activity [29].
4. palmitoylation modification, S nitrosylation modification
Palmitoylation refers to the process of covalently binding palmitic acid to cysteine residues through thioester bonds under the catalysis of palmitoyltransferase, which is a reversible post-translational lipid modification.
Cysteine C981 (the remaining sites C1176, 1178, and 1179) is the main palmitoylation modification site. After modification, it does not increase the number of Nav1.5, but it can improve the sodium channel opening characteristics and INaL, and enhance the excitability of cardiomyocytes. , And extend the duration of the action potential [30]. Due to the loss of cysteine sites, the C981F mutation can cause the steady-state inactivation of sodium channels, resulting in BrS [31]. Due to the introduction of cysteine residues, mutations including R34C, R1175C, F1473C and R1644C can induce LQTS, suggesting that the abnormal palmitoylation is involved in the occurrence of a variety of arrhythmias [30].
S nitrosation modification is a redox-based PTM, through which the nitric oxide (NO) group can be covalently bound to the cysteine sulfhydryl group to form S-nitrosothiol, thereby regulating Biological effects. Nav1.5 modified by S-nitrosylation can increase peak sodium current and INaL, and can be inhibited by caveolin 3 (CAV3) or N-ethylmaleimide. In type 9 LQTS and SIDS, CAV3 gene mutation can lead to loss of CAV3 function, causing excessive S nitrosylation modification of sodium channels [32]. At present, the specific cysteine residue positions of S-nitrosylation modification in Nav1.5 are still unclear.
5. Redox adjustment
The redox control system includes reactive oxygen species (ROS) production and antioxidant protein system. Abundant mitochondria, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase and NO synthase in myocardial tissue are important sources of ROS generation. After the methionine residue in Nav1.5 is oxidized by ROS, it can reduce the open rate and utilization of sodium channels, delay the resurrection time, and increase INaL.
In addition, ROS can activate the phosphorylation modification activity of the above kinases by oxidizing the methionine and cysteine groups in PKA, PKC, and CaMKⅡ. ROS can also mediate the lipid oxidative modification of Nav1.5, which can be regulated through a variety of indirect pathways. Electrophysiological properties of sodium channels [33, 34].
6. ubiquitination modification, ubiquitination modification
Ubiquitination modification is a process in which eukaryotic organisms covalently bind ubiquitin to target proteins through the enzyme-linked reaction of E1, E2, and E3 ubiquitinase, and play a role in regulating protein degradation, endocytosis, cell cycle and immune response. And other biological effects. Nav1.5 is the first ion channel reported to have ubiquitination modification in heart tissue.
The PY-motif at the carboxyl end can be recognized by the WW domain of the ubiquitin ligase Nedd4-2 and undergo ubiquitination modification. It can be degraded by the proteasome [35]. Inhibiting the expression of Nedd4-2 or using proteasome inhibitors MG132 or bepridil can significantly increase the number of channels and sodium current of Nav1.5.
However, there is still controversy about the ubiquitination modification site on the Nav1.5 channel. The homologous mutant mouse (SCN5A-p.Y1981N) constructed based on the PY motif mutation in patients with type 3 LQTS does not have electrical And abnormal sodium channel function [36].
Ubiquitin-like modification is a PTM mediated by ubiquitin-like protein SUMO. The process is similar to ubiquitination modification. SUMO is introduced into the substrate protein through the action of SUMO activating enzyme, binding enzyme and ligase. The ubiquitin-like modification of the lysine L442 site of Nav1.5 can promote the reopening of sodium channels in the late action potential and increase the INaL. Acute hypoxia stimulation can strengthen the level of modification and significantly extend the duration of the action potential [37] . The interaction between the SUMO binding enzyme UBC9 and Nedd4-2 is also an important mechanism for regulating the ubiquitination of Nav1.5. Inhibiting the activity of UBC9 can increase the expression of Nav1.5 and increase the sodium current density [38].
At present, there are no clinical reports of abnormal Nav1.5 ubiquitination and ubiquitination modification. Due to the low protein content and the diversity of ubiquitin chains, the existing mass spectrometry analysis cannot effectively target the precise target of the modification. The regulation of such modifications on cardiac function may be underestimated.
The voltage-gated sodium channel PTM as an important regulatory means to maintain the electrophysiological characteristics of cardiomyocytes is gradually being recognized and valued by researchers, which also provides new ideas for diagnosis and treatment of arrhythmias with negative genetic testing. With the support of high-efficiency mass spectrometry technology, exploring new PTM methods, PTM differences in pathophysiological states, and interactions between different PTMs will help clarify the relationship between the molecular characteristics of sodium channels and clinical diseases. The development of new drugs provides potential targets.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.