Cancer immunotherapy: Adoptive T cell (ATC) transfer therapy
- EPA Announces First-Ever Regulation for “Forever Chemicals” in Drinking Water
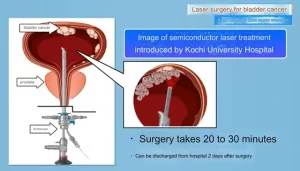
- Kochi University pioneers outpatient bladder cancer treatment using semiconductor lasers
- ASPEN 2024: Nutritional Therapy Strategies for Cancer and Critically Ill Patients
- Which lung cancer patients can benefit from neoadjuvant immunotherapy?
- Heme Iron Absorption: Why Meat Matters for Women’s Iron Needs
- “Miracle Weight-loss Drug” Semaglutide Is Not Always Effective
Cancer immunotherapy: Adoptive T cell (ATC) transfer therapy
Cancer immunotherapy: Adoptive T cell (ATC) transfer therapy. In recent years, adoptive T cell (ATC) therapy injects autologous or allogeneic T cells into cancer patients, which has shown considerable prospects. Southam et al. first proved the feasibility of this therapy.
In 1966, half of advanced cancer patients showed tumor regression after combined transplantation with patient-derived white blood cells and autologous tumor cells. Allogeneic hematopoietic stem cell transplantation for leukemia represents the first effective adoptive transfer method deployed clinically, and clinical improvement has been proven to be mediated by the anti-tumor response of T cell transplants.
01. ATC with tumor infiltrating lymphocytes
In the late 1980s, the National Cancer Institute took the lead in using tumor infiltrating lymphocytes (TIL) for ATC therapy to treat metastatic melanoma. The lymphocytes isolated from the cancer biopsy were greatly expanded with IL-2, and then a large amount of IL-2 was re-injected intravenously into the same patient.
The objective response rate was 34%; however, the median duration of the response was only 4 months, and few patients had a complete response. Later, the study of lymphatic removal before ATC treatment in 93 patients with metastatic melanoma was more successful. Twenty (22%) patients had complete tumor regression, and 19 patients were still in complete remission after 3 years of treatment.
High-throughput technology makes the screening and enrichment of neoantigen-specific TIL possible, and has recently shown promise in patients with metastatic breast cancer. In addition, knocking down the gene encoding the cytokine-inducible SH2 protein (Cish) (a negative regulator of TCR signaling) can enhance the anti-tumor response of ATC therapy in mouse models.
However, in order for TIL-based ATC therapy to trigger a durable response (Figure 5), effector T cells with anti-tumor activity must be present in the tumor, which is not the case for many cancer types. Other innovative ways to adjust T cell activity and proliferation may allow the development of more treatments.

Figure 5: Adoptive T cell therapy. Tumor infiltrating lymphocytes (TIL) are isolated from the patient’s tumor biopsy and expanded in vitro with IL-2. The TIL is then injected into patients who have experienced lymphatic depletion to provide a niche for the transferred TIL to expand, act as an effector cell, and generate immune memory. Since T cells are derived from tumors, it is assumed that most T cells can recognize tumor-associated antigens (TAA) or neoantigens.
02. Engineered lymphocytes for ATC
The challenges associated with the expansion of tumor-specific T cells in vitro have led to the development of TCR engineered lymphocytes (Figure 5). However, these cells are limited to responding to tumor antigens presented by MHC (also called human leukocyte antigens (HLA) in humans) rather than to surface antigens on tumor cells.
However, synthetic chimeric antigen receptors (CAR) can bypass Over MHC restriction and specific cytotoxicity directly act on target molecules on the surface of malignant cells. T cells isolated from patients (or allogeneic donors) are genetically modified to express CAR, and then expanded and injected into patients.
This overcomes this The problem that tumor cells often down-regulate MHC molecules makes it impossible for cells to present antigens to conventional T cells.
CAR contains an antigen binding domain, usually derived from the variable region of an antibody, connected to the signal domain of TCR and various costimulatory molecules (Figure 5). In view of the domain modularization of cell surface signal proteins, protein engineering can be used to assemble a mix and match of extracellular targeting domains and internal signal transduction domains. This provides many options for customizing CARs for specific tumors.
The first generation of CAR T cells only relied on the CD3ζ chain to simulate TCR signal transduction, but due to the limited T cell proliferation and cytokine production, this design was invalid in clinical trials. Subsequent generations of CAR T cells have been designed to include domains from CD28, CD40 ligands and other positive regulators of T cell activation to enhance in vivo activation and cytotoxicity. An engineered single-chain PD1 blocker also showed similar enhanced efficacy to second-generation CAR T cells with only the CD28 domain. Although CAR T cells are usually designed using retroviral transduction, recent work has used CRISPR-Cas9 technology. CRISPR-Cas9 can be used to directly edit the TCR germline sequence, which may lead to more uniform CAR T cell generation and ultimately better efficacy.
One limitation of the development of CAR T cell therapy is the need for unique tissue-restricted target antigens on the surface of tumor cells. For example, CAR T cells designed for the cell surface molecule CD19 expressed by all B cells have successfully treated B cell malignancies. The first clinical deployment of the second-generation CD19-specific CAR T cells resulted in a durable response to chronic lymphocytic leukemia. Other clinical trials of CD19-specific second-generation CAR T cells in B-cell acute lymphoblastic leukemia (B-ALL) resulted in remission of all B-ALL patients tested.
A follow-up report on B-ALL patients who participated in the clinical trial showed that 44 of 53 patients (83%) had complete disease remission, with a median follow-up time of 29 months. According to reports, patients with diffuse large B-cell lymphoma have achieved similar success, so the FDA approved these CAR-T cell therapies in 2017 for the treatment of B-cell malignancies.
The clinical success of CAR T-cell therapy in the treatment of B-ALL and diffuse large B-cell lymphoma is partly due to the targeting of CD19 antigen, which is an ideal candidate because it is highly expressed in certain B-cell malignancies and is Cell lineage. The cross-targeting of normal CD19+ B cells will not hinder treatment or cause serious side effects. However, even as an ideal target, loss of CD19 antigen is a common cause of treatment failure.
CD22 is another antigen commonly expressed by malignant cells in B-ALL, and has shown promise as a target for CAR T cell therapy in phase I trials. Other targets, especially tumor neoantigens, are currently being studied for hematological malignancies and solid tumors that do not express CD19. Based on promising preclinical and clinical data, B cell maturation antigen (BCMA) targeted CAR T cell therapy was approved by the FDA for multiple myeloma in 2021. However, due to the reported patient recurrence, research on other target antigens continues.
A recent preclinical study identified another target antigen, GPRC5D, whose efficacy and toxicity are comparable to those of BCMA targeting CAR T cell therapy. So far, CAR T cell therapy has only achieved certain success in solid tumors and innovative methods to improve the therapy are underway.
The recently identified pan-cancer target B7-H3 (also known as CD276) has been successful in multiple pediatric solid tumor models. In addition to directly acting as a cytolytic agent, CAR T cells can also target the harsh tumor microenvironment and restore depleted T cells. For example, a new generation of “armored” CAR T cells can be modified to produce IL-12, which can overcome the immunosuppression of Treg cells and bone marrow cells in the tumor environment, promote the cytolytic activity of CD8+ T cells, and enhance bone marrow cell recruitment and antigen presentation.
The efficacy of CAR T cells can also be enhanced by the co-expression of the chimeric cytokine receptor (4αβ), which stimulates the proliferation in response to IL-4, a cytokine that is usually abundant in the tumor microenvironment . Preliminary studies have shown that this method is suitable for CAR T cells targeting different tumor-associated antigens (TAA), and clinical trials for head and neck cancer are ongoing. In addition, overexpression of the transcription factor JUN showed resistance to CAR T cell depletion.
Overall, CAR T cells have successfully treated B-cell malignancies, and it will be exciting to continue research on this new treatment for refractory cancers.
03. Limitations and adverse effects of ATC
CART cell therapy can be toxic and affect many different organ systems with varying degrees of severity. Patients most often experience cytokine release syndrome (CRS) and neurotoxicity. CRS originates from the powerful activation and proliferation of CAR T cells in the body, and usually appears quickly after cell transfer. Symptoms are usually mild and flu-like, but can also be severe and life-threatening, including low blood pressure, high fever, capillary leakage, coagulopathy, and multiple system organ failure. Severe neurological events may also occur, such as CAR T cell-related encephalopathy syndrome, which is usually characterized by confusion and delirium, but is sometimes associated with seizures and cerebral edema. Glucocorticoids are the first-line treatment for mild CRS and CAR T cell-related encephalopathy syndrome.
Tocilizumab is a humanized anti-IL-6 antibody, a highly effective second-line therapeutic drug for the treatment of CRS caused by CART cell therapy [201]. Other side effects of CD19-specific CAR T-cell therapy include lymphopenia and hypogammaglobulinemia, which can be effectively controlled by intravenous immunoglobulin therapy, similar to the treatment received by patients with primary B-cell immunodeficiency. The mechanism behind these side effects is unclear, and further research may lead to ways to avoid or minimize toxicity. A new type of CRS mouse model developed recently shows that it is not mediated by CAR T cell-derived IL-6, but by receptor macrophages that secrete IL-6, IL-1, and nitric oxide. Therefore, IL-1 blockade represents a possible new intervention in the armament of CRS204.
In addition, a clinical study of low-affinity CD19-specific CAR T cells showed reduced toxicity and enhanced efficacy. Other efforts to reduce toxicity include designing CAR T cells with multiple receptor specificities and reducing the half-life of cytotoxicity through the use of mRNA-based methods that allow transient receptor expression or that can be activated by exogenous agents to perform The cloned suicide box deletes the injected cells.
The ATC method requires treatment design for a specific patient, its cost may be too high, patients have limited access to treatment, and manufacturing is challenging. In the United States, the direct costs of CAR T-cell therapy tisagenlecleucel and axicabtagene ciloleucel are US$475,000 and US$373,000 per patient, respectively. However, these values do not take into account the additional costs associated with treating serious adverse reactions common to CAR T-cell therapy, which is estimated to increase drug-related costs by US$30,000 or more209. Compared with CAR T cell therapy, the price of checkpoint blockade is approximately US$12,500 per month. Patient access to CAR T cell therapy also represents a major problem, because only a few laboratories are certified to produce CAR T cells, and only a few specialized tertiary medical centers can implement this therapy. Finally, the variability of CAR T cell manufacturing and the lack of standard practices can lead to different results.
Cancer immunotherapy focused on T cells has become a powerful tool in the fight against cancer. After years of basic scientific discoveries and subsequent clinical transformations, the ability to regulate the immune system to treat cancer has been clearly demonstrated. The successful cases of cancer immunotherapy that we have described highlight the inherent connection between basic scientific research and clinical practice. They also illustrate how a bench-to-bedside approach based on a solid foundation of science can successfully combat one of the most terrifying human diseases.
Sources from :
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.