Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities
Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities.
Most cancer-related deaths are due to metastasis, but our understanding of metastasis as an evolutionary, heterogeneous, systemic disease and how to effectively treat it is still emerging.
Metastasis requires a series of traits to spread, varying degrees of entry and exit from dormancy, and colonization in distant organs.
The success of these events is driven by clonal selection, the potential of metastatic cells to transition dynamically to different states, and their ability to exploit the immune environment.
Here, we review the key principles of metastasis and highlight new opportunities for developing more effective treatments for metastatic cancer.
Introduction
Metastasis refers to the growth of cancer cells in organs distant from their origin, representing the ultimate and most lethal manifestation of cancer. The vast majority of cancer patients die from metastatic disease rather than the primary tumor. Metastasis involves a series of biological events where cells from the primary tumor gradually acquire the ability to invade deeper tissues through the basement membrane, spread through the bloodstream, lymphatic vessels, or directly infiltrate adjacent structures, seed distant organs, and eventually proliferate in these organs to establish colonization.
These events are driven by tumor cells adopting different phenotypic cell states and their ability to absorb immune cells and stromal cells around them to support their growth and evade the immune system. While primary tumors can often be cured with local treatments such as surgery and radiation, metastatic cancer is a systemic disease affecting multiple organs, either by directly colonizing organs and impairing their function or by altering secretory profiles to change their metabolism, ultimately leading to death. Even within the same patient, the primary and metastatic diseases may respond very differently to systemic therapies. Clinical metastases that are apparent still largely remain incurable due to the acquired resistance of metastatic tumors to existing treatment modalities.
Technical advances in next-generation sequencing methods have been transformative for both basic cancer science and clinical oncology. They have accumulated tumor-specific expression patterns and tumor microenvironments by sequencing circulating tumor DNA (ctDNA) and circulating tumor cells (CTCs), tracking disease progression and patterns of resistance post-treatment, and elucidating the heterogeneity and clonality of primary and metastatic tumors.
These efforts together have yielded unprecedented developments in new biomarkers and drug targets, rapidly advancing our potential biological understanding of how metastatic cells hijack the host environment to ensure their survival. Here, we review existing paradigms of metastasis and emerging principles crucial for the dissemination, survival, and growth of metastatic cells, emphasizing recent discoveries and conceptual advances as well as their therapeutic implications for the treatment of metastatic cancer.
Stages of Metastasis
Metastasis can be divided into three stages—dissemination, dormancy, and colonization—during which cancer cells undergo a series of steps to invade tissues, survive in transit, and colonize organs, collectively referred to as the metastatic cascade.
During dissemination, tumor cells carrying oncogenic driver mutations invade deeper tissue layers through the basement membrane, gaining survival capabilities without the need for niche-specific growth factors.
Subsequently, they intravasate into proximal blood vessels or lymphatic vessels, eventually migrating across the endothelium and disrupting capillaries, spreading along nerve fibers, or directly locally infiltrating into adjacent spaces such as the peritoneal or pleural cavities to enter distant organs.
In circulation, CTCs undergo extensive attrition due to physical, oxidative, and immune stresses, a phenomenon confirmed in mouse models, inferring low numbers of CTCs from blood hours after resection of the primary tumor.
CTCs circulate as single cells or in clusters enriched in stem-like cancer cells, cloaked by platelets, neutrophils, or tumor-derived stromal cells, which can shield them from immune surveillance and endow CTC clusters with greater metastatic potential than single cells.
Upon arrival in distant organs, disseminated tumor cells (DTCs) face further elimination by active immune defenses, including high oxidative stress, lack of supportive growth factors or nutrients, and tissue-specific forms of macrophages, natural killer cells (NKs), infiltrating T cells, and other immune surveillance mechanisms.
Surviving DTCs can enter a reversible dormancy, during which they exit the cell cycle or enter a dynamic equilibrium of proliferation in response to immune elimination or other stromal inhibitory proliferation in the tumor microenvironment (TME), resulting in almost net metastatic growth.
Dissemination and dormancy are considered micro-metastatic diseases, as DTCs cannot be detected by clinical imaging and patients are unaware of subclinical disease.
Clinically overt metastases originate from successful metastatic initiating cells (MICs) that have adapted and absorbed their TME, ultimately enabling their growth and organ colonization.
The metastatic cascade represents a continuous cell and microenvironmental reprogramming and clonal selection of cancer cell subpopulations capable of withstanding selective microenvironmental pressures.
This leads to uncontrolled tumor growth, organ dysfunction, systemic organismal collapse, and ultimately death. This continuous change spans multiple domains and can be understood as the principles of metastasis.
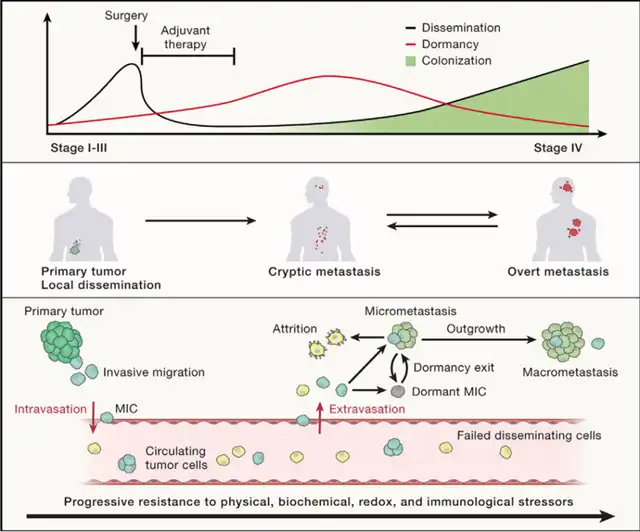
Figure 1. Stages of transfer
Metastasis consists of three stages: dispersal, dormancy, and colonization, which can coexist and overlap in time. MICs originate from primary tumors and acquire the ability to undergo invasive migration, then migrate and spread as CTCs individually or collectively through the blood or lymphatic vessels. Most CTCs are cleared due to physical, biochemical, and immune stressors. CTCs become trapped in the capillary beds of distant organs, extravasate as DTCs and migrate into the organ parenchyma to form nascent metastases. DTCs seed in organ-specific, perivascular niches Most are cleared by niche-specific or systemic immune defenses, but few survive, enter reversible growth arrest and immune-evading quiescence, acquire organ-specific growth adaptations, and exploit their TME to evade immune surveillance. Environmental triggers cause the mic to exit dormancy and form clinically detectable large metastases.
Principle of transfer
Metastatic cells require a large number of characteristics to undergo each step of the metastatic cascade. Some of these characteristics originate in the primary tumor and are the result of genetic mutations that activate oncogenes and disrupt tumor suppressor genes, allowing uncontrolled survival and proliferation, self-regeneration, migration, and invasion. But even with these oncogenic characteristics, the vast majority of cancer cells that leave the primary tumor cannot survive and form distant metastases. Metastasis is therefore a major evolutionary bottleneck.
Metastasis-specific traits emerge (1) by selection of these clones from the genetic heterogeneity present in the primary cancer cell population, or (2) by non-genetic dynamic adaptation of the cells, adapting the primary tumor to the needs of different metastatic stages.
Genetic Selection
The Cancer Evolution Paradigm
Systematic phylogenetic analyses of lineage relationships between matched primary and metastatic tumors reveal two broad evolutionary patterns of tumors during metastasis: in a linear model, metastatic cells spread from the primary tumor to the late stage, whereas in a parallel evolution model, cancer cells spread early, sharing a few mutations with the primary tumor and evolving separately from the primary tumor. Different modes of seeding (multi-clonal, mono-clonal, and metastatic reseeding) further increase the cellular heterogeneity of metastatic lesions. Genomic clonal evolution studies indicate early and late dissemination in different types of tumors and individuals. Recent multiplexed parallel lineage tracing work combining CRISPR-Cas9 editing with single-cell RNA sequencing has created an evolvable barcoding system, indicating that linear evolution and parallel evolution can also coexist.
Spatial Impact of the TME
Spatial genomics has furthered our understanding of intratumoral heterogeneity, subclonal variations, and the role of the local TME. The TME, composed of a variety of immune cells and stromal cells, interacts with and co-evolves with cancer cells, and can have both pro-tumor and anti-tumor effects. Spatial analysis of the TME in breast cancer patients shows that different cancer subclones are associated with varying degrees of local immune infiltration. In vivo spatial CRISPR screens in a mouse model of lung adenocarcinoma demonstrate that specific gene knockouts in tumor cells lead to characteristic changes in the immune environment and T cell infiltration. This suggests that tumor cells undergo local mutations under selection pressure, and conversely, clonal genetics may guide the establishment of local microenvironmental niches. Heterozygous loss of human leukocyte antigen class I allele genes is enriched in tumor subclones that undergo metastasis in various cancer types, while strong immunogenic neoantigens are preferentially lost in recurrent pancreatic cancer. Pre-existing lineage genetics can also influence the tendency to metastasize; recent studies suggest that lineage variations in the APOE gene alter the anti-tumor response of immune cells and result in different TME compositions and outcomes in a melanoma mouse model. The application of spatial and lineage tracing techniques in clinical samples and preclinical models holds promise for further revealing the mechanisms by which the TME influences MIC selection and how MICs utilize TME components to support their survival.
Copy Number Changes
While mutations are crucial for the initiation of tumors, large-scale genomic studies show that matched metastatic and primary tumors exhibit similar somatic mutation landscapes, and metastasis-specific somatic mutations cannot be easily identified. The discovery of new somatic mutations in the late stages of tumor progression often relates to treatment resistance. However, compared to some primary cancers, metastases exhibit copy number amplifications and chromosomal abnormalities but not universally. Myc amplification promotes metastasis by recruiting more tumor-associated macrophages (TAMs), leading to greater vascular invasion, providing evidence for the functional role of aneuploidy in driving metastasis. Chromosomal instability further promotes metastasis by increasing cytoplasmic DNA, resulting in activation of the cGAS-STING cytoplasmic DNA sensing pathway and downstream NFκB signaling. However, the extent to which aneuploidy plays a role in driving tumor progression in most cancers remains unresolved. Overall, genetic changes may allow for metastasis, but in most cases are insufficient to explain why some cancer cells metastasize while others do not.
Plasticity
Single-cell transcriptomic profiling of clinical samples and complex animal and patient-derived tumor models has revealed enormous transcriptional heterogeneity within and between tumors in advanced cancers, which cannot be explained by acquired genomic changes. Phenotypic plasticity, the ability to dynamically adapt to different stresses of the metastatic cascade and respond to changes in the TME, is therefore emerging as a primary hallmark of metastasis. Plasticity allows MIC cells to enter and exit stem-like states, undergo transdifferentiation, and dynamically adjust metabolism, oxidative stress, and immune stressors . Recent studies on metastatic mouse models suggest that cancer cells undergo multiple phenotypic transitions from the primary tumor to MICs and organ-specific metastases. In vivo lineage tracing and single-cell transcriptome analysis in a mouse model of lung adenocarcinoma show that selected high-plasticity primary tumor subclones are highly enriched in matched metastases, indicating that plasticity at the primary site is a prerequisite for metastasis. A key emerging theme is that the same molecules and pathways may play different roles in different environments during cancer progression, particularly in the context of primary tumor proliferation versus tumor dissemination and dormancy. Thus, designating cancer-relevant genes as oncogenes, from which tumor cells can benefit, or tumor suppressors, whose loss is always detrimental, often does not hold true in the dynamic context of metastatic plasticity.
Stress-responsive Regulation of Gene Expression
A wealth of research has led to the discovery of gene expression programs for discrete steps of the metastatic cascade. Epigenetic mechanisms, including DNA methylation, histone modifications, 3D chromatin organization, and non-coding RNAs, affect gene expression without altering the underlying DNA sequence, providing multiple possibilities for dynamic responses to environmental inputs. Activation of key transcription factors, including SOX10 and RUNX, has been shown to drive metastasis along an epigenetic continuum. Dynamic changes in histone modifications can alter chromatin accessibility and structure, with high-fat diets containing palmitic acid shown to drive metastasis in oral squamous cell carcinoma and melanoma models through the deposition of trimethylation of histone H3 lysine 4. MICs employ various stress response mechanisms to rapidly adapt their cellular state through post-transcriptional gene regulation, autophagy, and unfolded protein response, which are emerging as determinants of solid tumor metabolism and immune evasion plasticity. RNA-binding proteins coordinate cellular reprogramming and TME interactions during the metastatic process, regulating RNA modifications, mRNA splicing, localization, translation, stability, and degradation. Overexpression of the RNA N6-methyladenosine (m6A) reader YTHDF3 promotes the interaction and metastasis of cancer cells with brain endothelial cells and astrocytes by enhancing the translation of m6A-enriched transcripts. Cancer cells with lower levels of mitochondrial RNA cytosine-5 methylation (m5C) reduce the translation of mitochondrial-encoded oxidative phosphorylation complex members, blocking the switch from glycolysis to oxidative phosphorylation, a driver of metastasis in oral squamous cell carcinoma. Dynamic expression of specific tRNAs further promotes plasticity by regulating the translation dynamics of genes with different codon usage. Overall, these studies illustrate how cancer cells use epigenetic, transcriptional, and post-transcriptional landscape responses to evolve from evolutionary signals of organ-specific microenvironments.
Metabolic Adaptation
Although the metabolic shift of tumors to aerobic glycolysis (the Warburg effect) is a recognized hallmark of cancer, it is increasingly appreciated that the flux through metabolic pathways can be dynamically rewired during cancer progression. dtc can enable its metabolic adaptation to environmental stresses, including oxidative stress or nutrient availability, to survive during circulation and seeding distant organs. Metastatic cancer cells from different tumors have been shown to increase the uptake, synthesis, and utilization of lipids as a fuel source. Micrometastatic tumors can exploit autophagy and macropinocytosis to deal with nutrient deprivation or growth factor scarcity in the foreign organ environment. Overall, cancer cells rewire their metabolism during circulation and seeding, from a synthetic to a catabolic state and back to a synthetic state to support metastatic growth.
Reactive oxygen species (ROS) can induce cell ferroptosis. However, cancer cells can switch to an anti-ferroptotic state to increase metastasis, either by decreasing the synthesis of polyunsaturated ether phospholipids or by exposure to oleic acid in the lymph. In mouse pancreatic ductal adenocarcinoma, ROS initially restricts cancer-initiating stemness but later becomes a metabolic liability for metastatic cells. Therefore, the reversible effects of ROS enable the reciprocal conversion from proliferative phenotypes. Phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme of glucose-derived serine synthesis, has a similar context-dependent function, driving cancer proliferation, while PHDGH-dependent sialic acid synthesis and loss of integrin glycosylation increase metastatic dissemination. These studies highlight the importance of metabolic plasticity in cancer progression, with the same molecules and pathways playing different roles at discrete steps during proliferation and metastatic dissemination.
Co-option of Developmental and Regeneration Programs
While previous studies have identified individual genes and pathways associated with metastasis, modern systems biology approaches using unbiased profiling of the whole transcriptome, epigenome, and proteome of metastatic states are revealing convergent phenotypes. An emerging principle is that many cancers seem to co-opt common regulatory modules that activate pre-existing functionally related genes, thereby capturing many of the features required for metastasis. These gene networks are often required for the development or regeneration of the tissue of origin of the cancer, although cancer-specific novel gene programs can also contribute importantly .
Metastatic cells redeploy developmental and regeneration programs of normal embryonic development and wound healing. (A) In homeostasis, tissue-specific stem cells continuously generate transit-amplifying progenitor cells and mature differentiated cells. Upon tissue injury, differentiated epithelial cells dedifferentiate, re-entering a tissue-胎儿样, injury-associated transient progenitor state that can give rise to tissue stem cells, which then differentiate into differentiated cells, restoring the integrity of the epithelium. (B) Cell plasticity and fate are progressively restricted during embryonic development. In tissue injury, fate-restricted differentiated cells undergo a transient increase in plasticity. During cancer progression, cancer cells exploit developmental and regeneration progenitors to adapt to stress, although whether cells in overt metastasis are highly plastic or fate-restricted remains unclear. (C) Disseminated mic adopt highly plastic states. These states include hybrid EMT states, injury-associated transient progenitor-like states, or immune-逃避 dormancy states. During metastatic colonization, mic can regenerate phenotypically heterogeneous metastatic tumors, which can either enter dormancy or initiate tumor growth, re-entering a state similar to the primary tumor (elasticity), remain in a mic-like state (deformability), or undergo lineage plasticity to enter novel cell states not found in the primary tumor (transdifferentiation).
Development, Regeneration, and Metastasis
During embryonic development, the plasticity and fate of the progeny of the totipotent zygote are gradually restricted as organs mature. Most adult tissues contain a population of tissue-resident stem cells whose progeny can differentiate into all the different cell types of that tissue, but not into other tissues. Acquisition of oncogenic driver mutations in such homeostatic tissue stem cells leads to cancer, which are referred to as “cancer stem cells”. However, recent studies have suggested that tissue stem cells may not be a lineage-restricted population, but rather a metastable phenotypic state that many cells can enter and exit through plasticity, particularly during tissue regeneration following injury . In the lung, inflammatory injury induces basal cells to enter a mixed, injury-associated transient progenitor (DATP) state, with a gene expression signature that is hybrid of multiple normal lung epithelial cell lineages. DATPs in turn give rise to tissue-resident stem cell-like alveolar type II cells through plasticity, which then differentiate into different lung cell types to restore epithelial function and structure after injury. In mouse lung adenocarcinoma, a highly plastic cell state with multi-lineage differentiation potential has been identified as a precursor to metastasis, and reacquires a more primitive transcriptional program in mouse and human lung adenocarcinoma metastasis. Similarly, colorectal primary tumours are initiated by Lgr5+ cancer stem cells, but cells that spread from the invasive front of the tumour lose Lgr5 expression and instead acquire expression of new markers, including L1CAM and EMP1. L1CAM is not expressed by healthy intestinal epithelium, but is expressed by regenerating progenitors after injury, and is required for wound healing, metastatic initiation, and tumour regeneration after therapy. Established mouse colorectal metastases have been shown to revert from a Lgr5-low metastatic initiating state to a Lgr5+ state during metastatic outgrowth, although it is not clear to what extent this plasticity is retained in advanced human cancers.
Epithelial-mesenchymal transition (EMT) is the best-studied example of a developmental plasticity program that is co-opted in metastasis. This program involves a series of dynamic changes in cell organization, including loss of cell polarity and downregulation of epithelial cell adhesion molecules, leading to increased ability to migrate and invade neighbouring tissues. This is driven by the coordinated and dynamic regulatory function of the epithelial gene repressors ZEB and SNAIL transcription factors. EMT first occurs during embryonic gastrulation, as dtc acquire a migratory phenotype, and is recapitulated at the invasive front of primary tumours. Whether EMT is a prerequisite for metastasis has been controversial, as it has been difficult to prove in clinical samples, and some studies have suggested that EMT is not essential for metastasis but does contribute to chemoresistance. Recent data suggest that cancer cells often undergo incomplete or “partial” EMT.
Recent studies have shown that hybrid EMT states, co-expressing epithelial and mesenchymal markers, can drive metastasis in multiple cancer types, challenging the traditional view of EMT as a binary switch. As MICs acquire metastatic niche-specific growth capacity at distant sites, their resulting regenerating tumours can exhibit (1) plasticity, phenocopying the lineage hierarchy of the primary tumour, for example, through mesenchymal-to-epithelial transition (MET); (2) dedifferentiation, remaining trapped in a metastable MIC-like state; or (3) transdifferentiation, undergoing lineage plasticity to enter new cellular states with different tumour identities from the primary . The specific contribution of these three metastatic regeneration modes likely varies between individuals, tumour genotypes and tissues of origin, and remains to be determined in clinical metastasis.
Progressive immune escape
Evolution of the Tumor Microenvironment
DTCs must evade both tissue-resident and systemic immune attacks to successfully colonize distant organs. Significant evidence for the importance of immune surveillance of metastasis comes from cases of organ transplantation, where long-term recipients of kidney transplants for early melanoma cure develop widespread metastases due to micro-metastases in the kidney. Therefore, the increase of tumor-infiltrating lymphocytes in the primary tumor is a good prognostic biomarker for recurrence-free survival in colorectal cancer patients, while in experimental models, the depletion of T cells and NK cells increases metastasis. Immune checkpoint inhibitors (ICIs) that enhance anti-tumor immunity have profoundly changed the clinical practice of many metastatic cancers. Antibodies that block the interaction between cancer cells and T cells via PD-1, CTLA-4, and LAG3 can induce long-lasting responses in the clinic—unlike chemotherapy and targeted therapy—now the standard treatment for many solid tumors. Despite these successes, several tumor types do not respond to ICIs and are found to lack tumor T cell infiltration, either due to a lack of tumor antigens, antigen presentation defects, T cell activation defects, or T cell exclusion through immune-suppressive TME. Even for tumors initially responsive to ICIs, T cell exhaustion can occur, with chronic T cell stimulation leading to functional impairment. Importantly, ICIs seem to be more effective against primary tumors and micro-metastases than established large metastases. While adding PD-1 ICI pembrolizumab to chemotherapy showed no overall survival benefit in metastatic triple-negative breast cancer with low PD-L1 expression on tumor cells, adding pembrolizumab to early breast cancer resulted in significantly prolonged event-free survival regardless of PD-L1 status. Similar data for melanoma and lung cancer suggest that ICIs are more effective in the adjuvant/neoadjuvant setting than in advanced metastases. These clinical observations highlight the potential biological differences between primary tumors and metastases, as well as the progressive co-evolution of tumors with the immune-suppressive niche during tumor progression.
DTCs that infiltrate into distant organs first encounter macrophages and NK cells within the tissue, which further recruit local and circulating immune cells to combat the newly seeded metastasis. In response, cancer cells develop adaptive mechanisms to evade or suppress the immune response, limiting antigen recognition, increasing the expression of receptors that block adaptive immune systems, and secreting immune-modulating cytokines, extracellular vesicles, and growth factors. The TME includes T cells and B cells; NK cells; myeloid cells including TAMs, bone marrow-derived suppressor cells, dendritic cells, and neutrophils; stromal cells including cancer-associated fibroblasts, pericytes, and endothelial cells; and extracellular matrix (ECM) components, all of which coordinate these adaptations. The TME can have both tumor-promoting and tumor-suppressive effects simultaneously, but over time, its effects tend to become more tumor-promoting. Therapeutic targeting of the TME is increasingly being explored, either as a single agent or in combination with ICIs. Local and systemic immune suppression can occur before metastasis. Extracellular vesicles released from circulating cells from the primary tumor can induce the recruitment of bone marrow-derived immune cells and immune suppression reprogramming to the pre-metastatic organ site, where they form a “pre-metastatic niche” conducive to metastatic seeding. Signaling pathways that coordinate the functions of tissue-resident cells, recruited myeloid cells, and infiltrating tumor cells can further enhance the immune-suppressive environment that emerges during metastatic colonization.
The composition and co-selection of the TME are crucial for tumor growth and progression. The major components of the TME are the components of the innate and adaptive immune systems and stromal cells: tissue-resident and bone marrow-derived macrophages polarized into immune-suppressive TAMs, monocytes, bone marrow-derived suppressor cells, T cells, NK cells, dendritic cells, blood and lymphatic vessels, cancer-associated fibroblasts, and ECM components. In metastatic tumors, the immune cell composition of the TME and the expression characteristics of immune regulatory receptors are more immune-suppressive.
Lymph Node Metastasis
Metastatic immune suppression can also be orchestrated by cancer cells during transit. Clinically apparent tumor lymph node involvement is a strong prognostic biomarker for future metastatic disease, as reflected in clinical staging criteria. However, molecular reconstructions of clonal evolution show that lymph node and distant metastases mainly originate from independent subclones within the primary tumor. Recent studies suggest that lymph nodes are not passive staging posts in the metastatic process but critical sites for inducing systemic immune suppression. In a lymph node metastasis model, exposure to IFN-g in the lymph node induces the expression of PD-L1 and MHCI, promoting NK evasion and T cell suppression. Importantly, lymph node colonization promotes metastasis by inducing tumor-specific T regulatory cells, increasing PD-L1 expression on macrophages, and shifting dendritic cells from migratory to resident subtypes, altering systemic immune responses. Tumor transplant mice transplanted with white blood cells from lymph node tumor donors are more likely to develop lung metastases, suggesting that lymph node metastasis promotes metastasis by inducing tumor-specific immune tolerance. Further study of lymph node-dependent immune re-education could yield improved biomarkers and therapeutic targets to disrupt immune-suppressive circuits in metastatic cancer.
Dormancy
During dormancy, DTCs cannot form detectable macroscopic lesions, and patients only show clinical evidence of disease recurrence months or years later. Mouse models of early cancer suggest that DTCs can be found in every organ; however, DTCs eventually grow in a manner specific to a particular cancer only in specific tissues. Some cancers, such as estrogen receptor-positive breast cancer, can relapse distantly after surgery, up to 20 years after initial diagnosis, while others, including small-cell lung cancer, present with aggressive spread at diagnosis without measurable dormant periods. These observations suggest that the biological characteristics of dormancy entry and reawakening differ among different tumor types. Clinical dormancy reflects a dynamic balance of cellular dormancy, where DTCs enter a quiescent state through reversible growth arrest controlled by regulatory programs, tumor mass dormancy, random attempts at proliferation and colonization, interrupted by immune, physical, and metabolic barriers. Cellular plasticity is a key feature in maintaining dormancy. In a breast cancer model, early DTCs activate a stromal-like program associated with multicellularity, which coordinates dissemination and enables long-term dormancy controlled by the transcription factor ZFP281. Dormant DTCs maintain their state through epigenetic regulation, such as histone modification and enhanced DNA methylation, resulting in more suppressed chromatin states, transcription/post-transcriptional gene regulation, and activation of cell stress responses and autophagy. Proliferation of cancer cells is further controlled by growth inhibitory signals released by DTCs or TME components, including growth inhibitory signals and controls of collagen and laminin subtypes. Entry into cell quiescence has been shown to be functionally coupled with immune evasion, either through activation of the unfolded protein response, downregulation of NK ligands, or upregulation of the MacroH2A histone variant, coupled with resistance to IFN-g-induced cell death.
For Dormant Micro-Metastases
So far, drugs targeting the dissemination of metastases, such as matrix metalloproteinase inhibitors, have not been successful in clinical trials, consistent with clinical and preclinical evidence that metastasis starts early and may differ from the tumor’s origin. Typically, by the time the primary tumor is detected, disseminated tumor cells (DTCs) have already seeded in distant organs, making methods to block their spread ineffective. Perhaps the greatest opportunity to improve cancer prognosis lies in expanding clinical trials focused on dormant micro-metastases. As understanding of the biology of dormancy, plasticity, and molecular bases deepens, it becomes increasingly apparent that the markers, signaling pathways, and immune evasion strategies of micro-metastases differ from those of large metastases. However, the development of clinical drugs often proceeds by first testing new drugs in the late-stage metastatic environment, where tumors have developed resistance to most standard therapies. If successful here, trials advance to previously untreated metastases, then to adjuvant therapy for patients with no clinical metastases but a high risk of recurrence, where micro-metastases may exist. Therefore, many drugs that could be effective in treating micro-metastases, but not the more biologically invasive large metastases, fail to reach patients.
A key requirement for rapidly and cost-effectively advancing trials targeting micro-metastases is the establishment of actionable biomarkers for dormant micro-metastatic diseases to identify patients most likely to benefit from adjuvant therapy with the highest risk of recurrence from large metastases. In this regard, “liquid biopsy” ctDNA testing in blood is a promising biomarker for micro-metastases, but further improvements are needed in the sensitivity of this detection and the clinical operability of its results. Another proposed method for identifying patients with microscopic disease is the use of bone biopsies to identify DTCs in early-stage breast cancer patients. Future developments based on tissue protein or RNA analysis, using single-cell analysis of metastatic cells, detection of epigenetic modifications of ctDNA, or detection of CTCs, exosomes, immune cells, and cytokines, may provide avenues for the development of real-time predictive biomarkers. These methods will help stratify patients who continue to benefit from selected observation therapy and eradication of micro-metastatic diseases.
For Oligometastatic Diseases
Metastases can exhibit tumor-type-specific patterns of growth, spreading slowly or to a single organ site. Oligometastatic diseases are presumed to be an intermediate state of metastatic dissemination, where local ablative therapies can provide meaningful clinical benefits and prolong survival. In these cases, surgical resection or radiotherapy is often considered to reduce tumor burden, prolong life, and potentially cure the patient. Combined modality therapy in the form of surgery, radiotherapy, and chemotherapy is the standard treatment for soft tissue sarcomas, with systemic therapy alone showing limited efficacy. Surgical resection of liver metastases from colorectal cancer can cure 20% of patients and prolong the lives of patients with metastatic melanoma/skin or ocular melanoma and neuroendocrine tumors. Liver-directed therapies including hepatic arterial infusion chemotherapy, embolization, and radiofrequency ablation have shown survival benefits. Recent randomized controlled trials of local consolidative therapy for oligometastatic disease have shown prolonged progression-free survival and overall survival in breast cancer, prostate cancer, lung cancer, and other cancer patients. Alpha emitter radium-233 selectively binds to areas of increased bone turnover, demonstrating overall survival benefits in late-stage metastatic prostate cancer with bone metastases, suggesting that site-specific targeted therapy can affect subsequent widespread metastases, consistent with observations in mouse models. In conclusion, these studies indicate that widespread metastatic and oligometastatic diseases are clinically different, thus careful reconsideration is needed as local therapy typically does not improve survival rates for stage IV cancer patients. For patients with oligometastatic disease, there are currently few clinical tools to differentiate between diseases that may be organ-limited and those that are rapidly spreading with subclinical micro-metastases, which will soon become clinically apparent diseases in a short period of time. These two situations require different treatment approaches and have different outcomes, yet the need to understand the underlying biology and define biomarkers remains unmet.
Targeting Large Metastases
The plasticity of advanced metastatic disease poses a significant challenge to cancer treatment, as the ability to dynamically reprogram cell states theoretically allows almost any drug to become resistant. While single-cell maps of tumors have provided tremendous insights into tumor heterogeneity, more work is needed to determine the extent to which adaptive programs are shared among different tumor patients and host genetic or lifestyle factors. In principle, broad-spectrum therapies can be provided by targeting metastatic mediators that provide broad-spectrum therapeutic targets compared to patient-specific or broadly heterogeneous subclonal mechanisms.
However, research on developmental signaling pathways such as Wnt, TGFb, Notch, and Hedgehog has largely failed so far due to the high on-target toxicity of these pathways in tumors, environment-dependent pleiotropic effects, and feedback loops of these pathways. Other approaches focus on targeting metastatic cancer metabolism, such as inhibiting SLC6A8 creatine transporter in colorectal liver metastases, and describing metastasis-specific TMEs to overcome metastasis-specific immune-suppressive environments.
More preclinical research is needed to:
(1) focus on components that describe cancer-specific developmental and regenerative programs,
(2) identify targets that selectively regulate these pathways in a metastasis stage-specific environment of cancer cells and their evolving TMEs.
Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities
Cancer Metastasis: From Evolutionary Process to Therapeutic Opportunities
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



