The latest research progress of important microbiological journals
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
The latest research progress of important microbiological journals (20210412)
The latest research progress of important microbiological journals (20210412) .The CRISPR system uses RNA-guided nucleases to guide the sequence-specific destruction of the genome of movable genetic elements that regulate horizontal gene transfer (such as plasmaids and bacteriophages), thereby restricting the passage of microorganisms The degree to which this mechanism has evolved. According to the content of its cas gene, the CRISPR-Cas system can be divided into six different types (I-VI).
The mechanism of type III CRISPR-Cas immunization is the most complicated, and how this system affects the host has not been studied, so this is also the object of this article. The Rockefeller University Bacteriology Laboratory Charlie Y. Mo, Luciano A. Marraffini and others published an article entitled “Type III-A CRISPR immunity promotes mutagenesis of staphylococci” in Nature on April 7, 2021. This study showed that grapes The non-specific deoxyribonuclease activity of the Coccus III-A CRISPR-Cas system increases mutations in the host and accelerates the development of antibiotic resistance in Staphylococcus aureus and Staphylococcus epidermidis.
These mutations need to induce an SOS response to DNA damage (an induction response when DNA is damaged or DNA replication is blocked). The results indicate that the III-A CRISPR system can regulate the evolution of bacterial hosts by influencing the two mechanisms that produce genetic diversity.
Summary:
In order to enable bacteria to adapt to fluctuating environmental pressures, horizontal gene transfer and mutation are the two main driving forces of microbial evolution. Aggregated, regularly spaced short palindromic repeats (CRISPR) systems use RNA-guided nucleases to guide the control of movable genetic elements that regulate horizontal gene transfer (such as conjugated plasmin (plasmids) and bacteriophages). The genome undergoes sequence-specific destruction, which limits the degree to which microorganisms can evolve through this mechanism.
Research on a subset of the CRISPR system found non-specific degradation of DNA; however, whether and how this feature affects the host has not been studied. This study shows that the non-specific deoxyribonuclease activity of the Staphylococcus III-A CRISPR-Cas system increases mutations in the host and accelerates the development of antibiotic resistance in Staphylococcus aureus and Staphylococcus epidermidis.
These mutations need to induce an SOS response to DNA damage (an induction response when DNA is damaged or DNA replication is blocked). The results show that the III-A CRISPR system can regulate the evolution of bacterial hosts by influencing the two mechanisms that produce genetic diversity.
Type III-A CRISPR immunity promotes mutagenesis of staphylococci
Journal: Nature
IF: 42.778
Publication time: 2021.4.7
Corresponding author: Charlie Y. Mo, Luciano A. Marraffini
Corresponding Author Unit: Bacteriology Laboratory, Rockefeller University
DOI number: 10.1038/s41586-021-03440-3
Original link:
https://www.nature.com/articles/s41586-021-03440-3
Opinion | Nature Medicine: A scientific framework for the microbiome in public health
Wendy S. Garrett, Curtis Huttenhower and others in the Microbiology Group of the Chen Zengxi School of Public Health, Harvard University, USA published a viewpoint article entitled “A framework for microbiome science in public health” in Nature Medicine on April 5, 2021. This study discusses how microbiome science is applied and integrated into public health research and practice. It also explains the necessity of decision-making and supervision in related fields.
It points out:
(1) The combination of microbiome science and public health will Contribute to the discovery of new biomarkers, therapies or molecular mechanisms;
(2) Microbiome epidemiology has many of the same potentials and methods as other forms of molecular epidemiology;
(3) It is necessary to improve research related basic equipment and Application resources;
(4) Microbiome science has great application prospects in public health, such as nutrition and health care, developmental aging, chronic diseases, and infectious disease control;
(5) Microbiome science in clinical trials, microbial products, and public health The education and training of personnel, as well as policies and regulations, also play a pivotal role.
The viewpoints in this article have important guiding significance for the integrated research of the microbiome and public health and the development of related fields.
Abstract: The science of the human microbiome has developed rapidly and has reached the scale of increasing integration of basic biology, clinical transformation and population health. Therefore, public health researchers, practitioners, and decision makers can now take advantage of current and future microbiome-based opportunities and best practices to take concrete actions. Here, we provide an outline of considerations for research, education, interpretation, and scientific communication about the human microbiome and public health. This includes population-scale microbiome research design guidelines; necessary physical platforms and analysis methods; integration into public health fields such as epidemiology, nutritional science, chronic diseases, and global and environmental health; entrepreneurship and technology transfer, and educational courses. Especially in the near future, there will not only be opportunities to incorporate microbiome-based technologies into public health practices, but there will also be an increasing need for decision-making and supervision around related areas, such as prebiotics and probiotic supplements, new live cell therapies and Fecal microbiota transplantation, etc.
A framework for microbiome science in public health
Journal: Nature Medicine
IF: 36.13
Publication time: 2021.4.5
Corresponding author: Wendy S. Garrett, Curtis Huttenhower
Corresponding Author Unit: Microbiology Group, Chen Zengxi School of Public Health, Harvard University, USA
DOI number: 10.1038/s41591-021-01258-0
Original link:
https://www.nature.com/articles/s41591-021-01258-0
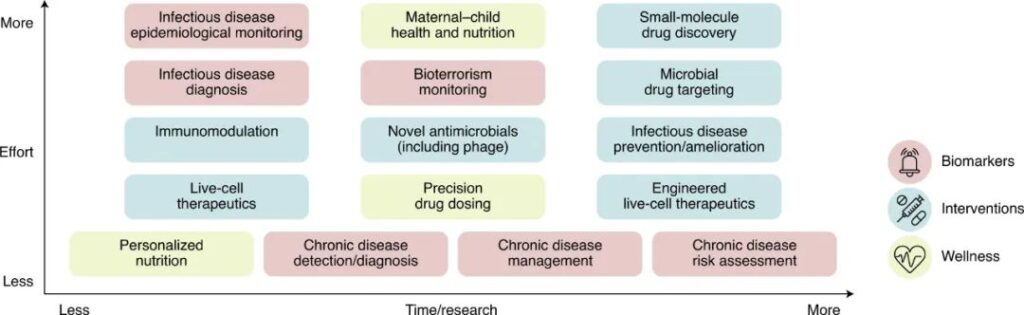
Figure Opportunities for the development of microbiome science in public health
Research | Nature Medicine: The role of integrated microbiome in acute exacerbation of bronchiectasis
The main feature of Bronchiectasis is repeated infection and inflammation of microorganisms. Studies have shown that bacterial, viral and fungal communities in the respiratory tract are importantly related to the clinical prognosis of patients with bronchiectasis. Although specific pathogens are involved in the deterioration of bronchiectasis, previous studies on the bacterial microbiome have shown that the actual changes in the bacterial microbiome are minimal during the analysis of dominant groups or differential indicators based on deterioration. Therefore, we have The role of is not fully understood. On April 5th, Sanjay H. Chotirmall and others from the Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore, published an article entitled “Integrative microbiomics in bronchiectasis exacerbations” in Nature Medicine. Through a network analysis of the respiratory tract microbiota of patients at risk of bronchiectasis, a systematic Clarified the basic characteristics of microbial co-occurrence networks of bacteria, viruses and fungi in patients at high risk of deterioration, and revealed the influence of antagonism between microorganisms on the disease process. Integrating the network characteristics of the microbiome and the interaction information of microorganisms can improve the effect of clinical prediction models. The results of this study provide ideas for understanding the relationship between bronchiectasis and the microbial community of the respiratory tract, and at the same time provide guidance for new strategies in the use of antibiotics, that is, antibiotic treatment should target the interaction network of microorganisms, rather than acting alone microorganism.
Abstract: Bronchiectasis is a progressive chronic airway disease, which is mainly characterized by microbial colonization and infection. We propose a multi-biome method to integrate the bacterial, viral and fungal communities of patients with bronchiectasis through weighted similarity network fusion (https://integrative-microbiomics. ntu.edu.sg). The microbial co-occurrence networks of patients with the highest risk of deterioration have lower complexity, decreased diversity, and a higher degree of antagonistic interactions in the respiratory microbiome (microbiome). In addition, longitudinal interactome dynamics reveals the antagonism of microorganisms during the deterioration process, which solves the problem of subsequent treatment in an otherwise stable multi-biological community. The evaluation of the Pseudomonas interactome shows that the interaction network, rather than the individual abundance, is associated with the risk of exacerbation, and the introduction of microbial interaction data improves the clinical prediction model. Shotgun metagenomic sequencing in an independent cohort validated the multibiome interaction detected in targeted analysis and confirmed its correlation with deterioration. Integrated microbiology captures the interaction of microorganisms to determine the risk of deterioration, which cannot be determined by the study of a single type of microorganism. Antibiotic use strategies should probably target interaction networks rather than individual microorganisms, which provides a new way to understand respiratory infections.
Keywords: bronchiectasis, integrated microbiome, multi-microbiome, interactome, network analysis
Integrative microbiomics in bronchiectasis exacerbations
Journal: Nature Medicine
IF: 36.130
Publication time: 2021.4.5
Corresponding author: Sanjay H. Chotirmall
Corresponding Author Unit: Lee Kong Chian School of Medicine, Nanyang Technological University, Singapore
DOI number: 10.1038/s41591-021-01289-7
Original link:
https://www.nature.com/articles/s41591-021-01289-7
Scientific research | Nature Plants: Phytoflavonoid secretions improve the nitrogen deficiency traits of corn by accumulating oxalobacter in the rhizosphere
Professor Chen-ping of Resources and Environment, Southwest University joint German Crop Functional Genomics Laboratory of the University of Bonn Professor Frank Hochholdinger (first author: Yu Peng & Xiaoming) on April 8, 2021 speech entitled Nature Plants “Plant flavones enrich rhizosphere Oxalobacteraceae to Improve maize performance under nitrogen deprivation” article. The research respectively passed: transcriptome sequencing, 16S and ITS amplicon sequencing on the longitudinal root region of 20 maize inbred line materials with different genetic backgrounds; metagenomic sequencing, rhizosphere transplantation test on representative inbred lines, Inoculation experiments of different soil isolates; targeted metabolite analysis of corn rhizosphere (exudates), stable 14C label tracing experiment, and the feedback regulation mechanism between corn root structure and specific functional microbial communities under nitrogen deficiency conditions . Studies have shown that under conditions of nitrogen deficiency, corn will affect its rhizosphere microflora through specific flavonoid secretions, which in turn affects the host’s lateral root development and nutrient absorption. This research reveals the genetic basis of the interaction between root structure and specific microbial groups in the rhizosphere. This work provides new ideas for how to make full use of the benign interaction of crop roots and rhizosphere microbes, thereby improving the absorption and utilization efficiency of soil nutrients by crops. The research approach and thinking.
Abstract:
The benign interaction between plant roots and rhizosphere microorganisms is essential for crop growth. However, for corn crops, the feedback regulation mechanism between the root structure and the rhizosphere microbial community is still unclear.
In this study, we found that the functional characteristics (transcriptome) of the longitudinal development area of the corn root system are related to the specific microbial diversity; secondly, the flavonoid root exudates can drive the corn rhizosphere to accumulate oxalobacter, thereby promoting corn growth and nitrogen In the end, under nitrogen-deficiency conditions, it is the mutant LRT1 that coordinates the interaction between the corn roots and the flavonoid-dependent rhizobacillus oxalicum, which itself can induce the formation of lateral roots.
In summary, our research revealed that the interaction between the root structure and specific microbial groups in the rhizosphere of maize can improve the characteristics of maize plants under nitrogen deficiency. This discovery may open up new ways to develop high-yield and high-nutrient crops by adjusting the interaction between crops and beneficial soil microorganisms.
Plant flavones enrich rhizosphere Oxalobacteraceae to improve maize performance under nitrogen deprivation
Journal: Nature Plants
IF: 13.256
Issuing time: 2021.04.08
Corresponding author: Chen-ping & Frank Hochholdinger
Corresponding Author Unit: School of Resources and Environment, Southwest University
DOI number: 10.1038/s41477-021-00897-y
Original link:
https://www.nature.com/articles/s41477-021-00897-y
ISME Journal
Research | The ISME Journal: Microbiome assembly and genetic determinants transfer in the presence of sulfonamides
Zhang Tong’s team from the Environmental Microbial Engineering and Biotechnology Laboratory of the Department of Civil Engineering of the University of Hong Kong published an article entitled “Microbiome assembly for sulfonamide subsistence and the transfer of genetic determinants” in The ISME Journal on April 5, 2021. The article studied sulfonamides. A combination of drug-like microorganisms, using wastewater microbiota from six regional pools as inoculants, and using sulfa antibiotics as the sole carbon source to assemble them. It spans the hierarchical structure of the microbial community: from communities and individuals to pathways and genes, and proposes primary degrader-mediated determinism for the observed patterns. And by using short-read and long-read sequencing, the study isolated complete or nearly complete genomes and mobiles (collections of all mobile elements) from a single isolate that can maintain sulfadiazine. Reveals previously undetected polymorphisms associated with sulfa metabolism gene clusters. More importantly, the special ability of sulfa drugs to maintain survival is shown by evolutionary conservation, showing limited spread outside the boundaries of Micrococcaceae. This research opens up new possibilities for the environmental application of bacteria in engineering specialty.
Abstract:
The presence of antibiotics in bacteria represents an alternative resistance mechanism, and paradoxically, it is also a treatment for environmental resistance. Antibiotics exist bacteria can detoxify the environment contaminated by antibiotics and prevent the development of antibiotic resistance in the environment. However, the lack of a mechanistic and predictive understanding of the assembly of functional microbiomes has hindered the progress of effective in situ engineering of antimicrobial bacteria.
By using sulfadiazine as a single restrictive source to manipulate the wastewater microbiota from top to bottom, we monitored the ecological selection process that forces the wastewater microbiota to survive highly efficient sulfadiazine. We found that in different initial microbial populations, the combination of community levels selected the same three families. We used linear models to further analyze the combination mode.
A detailed examination of the sulfa metabolism gene clusters in the individual genomes of isolates and collective metagenomics revealed limited metastatic potential outside the lineage boundaries of the Micrococcaceae family. Our results open up new possibilities for the environmental application of engineering specialty bacteria.
Original name: Microbiome assembly for sulfonamide subsistence and the transfer of genetic determinants
Translation: Microbiome assembly and genetic determinants transfer in the presence of sulfa
Journal: The ISME Journal
IF: 9.180
Issuing time: 2021.04.05
Corresponding author: Zhang Tong
Corresponding Author Unit: Laboratory of Environmental Microbiology Engineering and Biotechnology, Department of Civil Engineering, The University of Hong Kong
DOI number: 10.1038/s41396-021-00969-z
Original link:
https://www.nature.com/articles/s41396-021-00969-z
Critical Reviews in Microbiology
Review | Critical Reviews in Microbiology: Intestinal microbe-microRNA interaction regulates host gene expression to treat human diseases
Vibha Rani, Transcriptome Laboratory, Center for Emerging Diseases, Department of Biotechnology, Jaypee Institute of Information Technology, India, published a titled “Modulating host gene expression via gut microbiome–microRNA interplay to treat human” in Critical Reviews in Microbiology on April 6, 2021. A review of diseases. This review reviews the basic principles of miRNA technology and its interaction with intestinal flora. It also brings together its related important research progress and outstanding findings. The main three aspects are explained layer by layer, starting with the introduction of the basic role of miRNA, including miRNA and its mediated gene regulation, and microRNA participating in the development of the host immune system. Secondly, it introduces the relationship between the flora and human health, including the control of host health by intestinal microbes. Finally, the interaction between miRNA and flora is introduced, mainly from the impact of microbial-miRNA interaction on host gene expression and intestinal immunity, and the impact of intestinal flora-microRNA interaction in human diseases, including inflammatory enteritis ( IBD), liver disease, cardiovascular disease (CVD), nervous system disease, cancer, and intestinal miRNA-based treatments. Explore the interaction between miRNAs and flora, and emphasize its ability to regulate host gene expression, thereby formulating potential therapeutic intervention strategies for various human diseases.
Abstract:
The human gastrointestinal tract is involved in maintaining the balance of the intestinal environment and the habitat of trillions of microorganisms. Microbial disorders in the intestine can cause a series of pathogenic and autoimmune diseases. Although the mechanisms by which the microbiota regulates human health are multifaceted, the metabolites released from the dietary supplements they ingest play a crucial role in bidirectionally regulating the expression of small ribonucleic acids (miRNAs).
miRNAs are small endogenous non-coding RNAs (ncRNAs) that have been confirmed to participate in the interaction of microbial regions and regulate the expression of host genes. This review focuses on the key principles, regulation, and correlation of miRNAs with the intestinal microbiota to affect the expression of host genes in various human diseases; it brings together important recent discoveries centered on miRNA-microbes. These findings The interaction with other organs in diseases along different axes of the intestine unfolds.
This article also attempts to focus on the intestinal-oriented miRNA therapy approach, using basic dietary supplements to regulate the abnormal expression of host genes in diseases, providing a new perspective for the realization of economic treatment.
Keywords: MicroRNA; intestinal microflora; gene regulation; human diseases; intestinal therapy
Original name: Modulating host gene expression via gut microbiome–microRNA interplay to treat human diseases
Translation: gut microbe-microRNA interaction regulates host gene expression to treat human diseases
Journal: Critical Reviews in Microbiology
IF: 7.349
Issuing time: 2021.04.06
Corresponding author: Vibha Rani
Corresponding Author Unit: Transcriptome Laboratory, Center for Emerging Diseases, Department of Biotechnology, Jaypee Institute of Information Technology, India
DOI number: 10.1080/1040841X.2021.1907739
Original link:
https://doi.org/10.1080/1040841X.2021.1907739
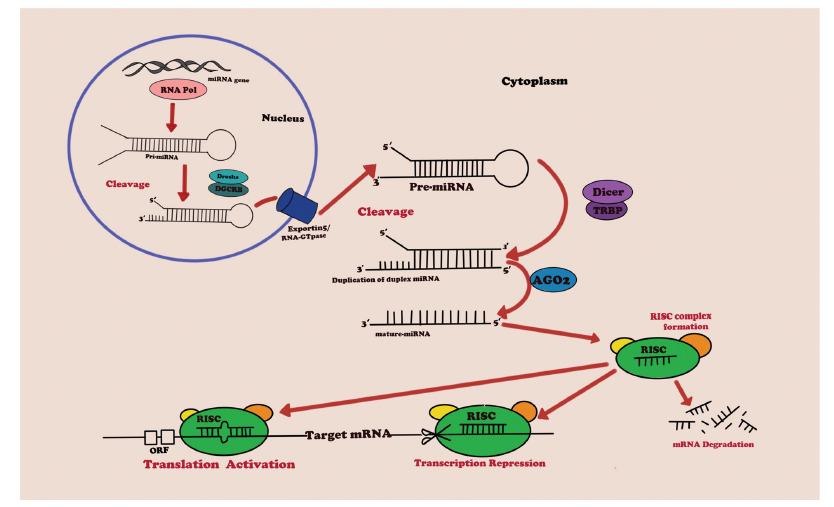
Figure Typical pathways and mechanism of miRNA biogenesis
mSystems
Research | mSystems: Revealing system symbiosis and microbial-assisted hybrid degradation from the gut bacterial genome of wasp
Seth R. Bordenstein, Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee, USA, published a titled “Genomes of Gut Bacteria from Nasonia Wasps Shed Light on Phylosymbiosis and Microbe-Assisted Hybrid Breakdown” at mSystems on April 6, 2021 In the article, this study used the intestinal bacteria in the wasp body to sequence the whole genome, and compared and analyzed the most abundant intestinal bacteria in the golden wasp larva, Providencia rethei and Proteus mirabilis. It was found that under the same culture conditions, Proteus mirabilis was significantly better than Providencia reesei. The gut microbiota in the larvae of golden bee was the most similar. The dominant species in the hybrids-Proteus mirabilis was in the parent species. Finally, it is concluded that the holographic biological interaction between the symbiotic bacteria and the host in the wasp microbial community is the basis of system symbiosis and hybrid degradation, which provides an important reference for studying system symbiosis and microbial-assisted hybrid degradation. significance.
Abstract:
Phylosymbiosis has a cross-system trend. This is because the relationship between the microbial communities reproduces the host phylogeny. In the parasitic wasp of Nasonia, system symbiosis runs through its entire development process. System symbiosis not only distinguishes gender, but also benefits the development and survival of the host. In addition, when a rare Proteus (Proteus) in the microbiota becomes a dominant population, the microbiota in the hybrid will change.
The larvae will completely die from bacterium-assisted lethality and reproductive isolation between species after hybridization. Two important questions to understand the systemic symbiosis of hybrids and the lethality of auxiliary bacteria are: (i) Is the genome of the golden bee different from other isolated animals; (ii) Is the genome of the hybrid bacteria the same as the parent? In this article, we report how to cultivate bacteria, sequence their whole genome, and compare and analyze the larvae (Nasonia larvae), Providencia rettgeri (Providencia rettgeri) and Proteus mirabilis ( Proteus mirabilis) is the most abundant intestinal bacteria.
The characteristics of the new isolate indicate that the biofilm formed by Proteus mirabilis is stronger than that of Providencia rethei. When grown together under the same culture conditions, Proteus mirabilis is significantly better than Providencia rethei . The genomes of Providencia renzi from the golden bee are similar to each other and are more different from the pathogenic bacteria related to humans. Proteus mirabilis and its hybrid progeny in Nasonia vitripennis (Nasonia vitripennis) and Nasonia giraulti (Nasonia giraulti) are almost identical, but they are relatively different from artificially isolated bacteria.
These results indicate that the gut microbiota in the larvae of the golden bee is the most similar, and the dominant Proteus mirabilis in the hybrid species exists in the parent species. Holobiont interactions between the symbiotic bacteria and the host in the wasp microbiota are the basis of system symbiosis and hybrid degradation.
Keywords: Proteus, Providencia, Golden Bee, Gut Bacteria, Microbiome, Bacteriophage
Genomes of Gut Bacteria from Nasonia Wasps Shed Light on Phylosymbiosis and Microbe-Assisted Hybrid Breakdown
Journal: mSystems
IF: 6.633
Issuing time: 2021.04.06
Corresponding author: Seth R. Bordenstein
Corresponding Author Unit: Department of Biological Sciences, Vanderbilt University, Nashville, Tennessee, USA
DOI number: 10.1128/mSystems.01342-20
Original link:
https://msystems.asm.org/content/6/2/e01342-20
Environmental Microbiology
Research | Environ. Microbiol.: Termite mound reduces soil microbial diversity by filtering rare microbial groups
Hang-Wei Hu, School of Veterinary and Agricultural Sciences, University of Melbourne, Australia, published an article entitled “Termite mounds reduce soil microbial diversity by filtering rare microbial taxa” in Environmental Microbiology on April 5, 2021. Because dominant microbial groups generally have higher potential in resource utilization, competition and resistance than rare microbial groups. Therefore, this study speculates that due to the more intense microbial competition in the termite mound, the relative abundance of rare soil microbial groups in the termite mound may decrease. In order to verify this hypothesis, this study used amplicon sequencing technology to analyze the diversity and composition of microbial communities in 134 termite mounds in 16 locations in northern Australia.
A large-scale investigation was conducted. The results show that the process of termite nesting may enrich the dominant taxa in the soil and filter the rare taxa at the same time. Some eutrophic groups (for example, Actinobacteria, Bacteroidetes) can use the higher nutrients in termite mounds to expand their populations, while oligotrophic groups may lose competitiveness, resulting in a decrease in their abundance. The results of this study deepen the understanding of how termite nesting processes shape soil microbial communities, and are of great significance for better predicting the ecological functions of termite mounds in changing environments.
Abstract:
Termites are insects that are ubiquitous in tropical and subtropical habitats. Some termites build huge nests (“mounds”). These nests greatly promote the heterogeneity of the substrate by changing the characteristics of the soil. However, the role of termite nesting process in regulating the distribution and diversity of soil microbial communities is still poorly understood, which brings uncertainty to the prediction of termite mound ecosystem functions in a constantly changing environment.
Therefore, this study used amplicon sequencing technology to investigate 134 termite mounds within a range of more than 1500 km in northern Australia, and found that the soil microbial diversity and community composition of termite mounds are significantly different from ordinary loose soil. Compared with loose soil, termite nesting process reduces soil microbial diversity and the relative abundance of rare taxa. The habitat niche width of the rare taxa is narrower than that of the dominant taxa, and may be more easily filtered by potential fierce microbial competition during the nesting process.
This study further proves that the pH change caused by termite nesting is the main driving factor shaping the characteristics of the microbial community in the termite mound. In conclusion, this study provides new evidence that termite nesting is an important process for regulating soil microbial diversity, thereby improving the understanding of the function of termite mounds.
Termite mounds reduce soil microbial diversity by filtering rare microbial taxa
Journal: Environmental Microbiology
IF: 4.933
Issuing time: 2021.04.05
Corresponding author: Hang-Wei Hu
Corresponding author unit: School of Veterinary and Agricultural Sciences, University of Melbourne, Australia
DOI number: 10.1111/1462-2920.15507
Original link:
https://sfamjournals.onlinelibrary.wiley.com/doi/abs/10.1111/1462-2920.15507
Research | Environmental Microbiology: Environmentally soluble DNA provides meaningful biological information for microbial community structure
Department of Physiology, Genetics and Microbiology, University of Alicante, Spain, Fernando Santos et al. published an article entitled “Environmental dissolved DNA harbors meaningful biological information on microbial community structure” in Environmental Microbiology on April 5, 2021. Intracellular DNA (Extracellular DNA, eDNA) and viral DNA have received extensive attention from microbial molecular ecologists. The study of soluble DNA fragments can help clarify the dynamics and evolution of ecosystems, and is of great significance to ecological research. However, for the separation of soluble DNA, care must be taken as much as possible, and the operation of each sample must start with the most optimized method. For high saline samples, filtration is the most suitable method to remove cells, which can avoid cell damage and unreproducible output. In this study, the dissolved DNA (dDNA) from a high-salt environment revealed that the dDNA is composed of cell and viral DNA, and its ratio is different from those found in cells or in the viral DNA library. It is nanometer The virus activity of grade archaea provides clues. In addition, experimental evidence supports the new function of dDNA as an ultraviolet protective agent.
Abstract:
Extracellular DNA (Extracellular DNA, eDNA) contains all DNA molecules outside the cell. This component of the microbial ecosystem may be a source of nutritional and genetic information. According to reports, the high-salt environment is one of the environments with the highest eDNA concentration in the natural ecosystem, which is believed to be due to the physical and chemical protective effects of salt and the high virus abundance. In this study, we compared two methods of extracting dissolved DNA (dDNA, instead of eDNA that also contains DNA from free viral particles) from water samples of the solar crystallization salt pond (CR30), namely centrifugation and filtration.
For the first time, crystalline dDNA fragments were characterized, and cell and viral metagenomics were compared at the same site. Due to cell lysis, high-speed centrifugation affects the concentration and composition of CR30 dDNA, thus emphasizing that protocol optimization is the first step in dDNA research. Crystalline dDNA, lower than the previously reported dDNA concentration in salt water anoxic sediments, originated from viruses and cells, is enriched in archaeal DNA, and is different from dDNA derived from a combination of cells in the same sample Classification composition. Bioinformatics analysis shows that nanoarchaea viruses are the cause of these differences.
Keywords: eDNA, dDNA, high salt, nano-archaea, nano-virus, salt algae
Environmental dissolved DNA harbors meaningful biological information on microbial community structure
Journal: Environmental Microbiology
IF: 4.933
Publication time: 2021.4.5
Corresponding author: Fernando Santos
Corresponding Author Unit: Department of Physiology, Genetics and Microbiology, University of Alicante, Spain
DOI number: 10.1111/1426-2920.15510
Original link:
https://sfamjournals.onlinelibrary.wiley.com/doi/epdf/10.1111/1462-2920.15510
Microbiological Research
Research | Microbiological Research: Plant Microbiology for Sustainable Agriculture and Food Security: Opportunities, Challenges and Solutions
Brajesh Kumar Singh and others from the Institute of Environment and Sustainable Development, Banaras Hindu University, Varanasi, Uttar Pradesh, India, published a titled “Phytomicrobiome for promoting sustainable agriculture and food security: opportunities” in Microbiological Research on April 5, 2021. , challenges, and solutions” article, this article describes the functional characteristics of plant microbiota, focusing on cultivable microorganisms to promote sustainable agriculture. It mainly includes: (1) the colonization process explored, (2) emphasizes the mechanism and functional characteristics of the plant microbiome, and (3) describes new methods and future prospects for the challenge of the plant microbiome.
Abstract:
Ensuring food security in an environmentally sustainable way is a global challenge. To achieve this goal, agricultural productivity needs to be increased by 70% under increasingly severe weather conditions without further damaging the quality of the environment (such as reducing the use of agrochemicals). Most governments and intergovernmental agencies have emphasized the need for alternative methods of using natural resources to solve this problem. The use of beneficial plant microbiota (that is, microorganisms closely related to plant tissues) is considered to be one of the feasible solutions to the dual challenges of food security and environmental sustainability.
A variety of important microorganisms are found in different parts of plants, namely roots, stems, leaves, seeds and flowers. They play an important role in plant health, development and productivity, and can directly contribute to improving the quality of food production. And quantity. Plant microbiota can also increase productivity by improving resource utilization efficiency and resilience to biological and non-biological stress. In this article, we explored the role of plant microbiota in plant health and how to use the functional properties of microbiota to increase agricultural productivity in an environmentally friendly way. However, there are still major technical and transformational challenges, such as the inconsistent efficacy of microbial products under field conditions, and the lack of tools to manipulate microorganisms in situ.
We have proposed some approaches, such as the use of consortia, synthetic communities (as opposed to individual isolates), endophytic microorganisms, and better formulation techniques. These approaches require a system-based approach to realize the potential of plant microbiota in promoting food security. We believe that if these technology and transformation limitations can be systematically addressed, plant microbiota can make a huge contribution to the sustainable growth of agricultural productivity and food security. In recent years, some progress has been made in the field of plant microbes, but there are still many knowledge gaps that require priority attention, including:
(a) understanding of the basic process of plant tissue microbial colonization,
(b) cultivation in the rhizosphere and endophyte Characterization of the biochemical/molecular signals of beneficial microorganisms;
(c) Identify the core and central microbiota associated with plants and how they are affected by agronomic measures and climate change.
If these gaps are systematically resolved, plant microbiology methods may provide tools for future transformational growth in agricultural productivity and environmental sustainability. Under different environmental stresses in the future, plant microbiota may become more effective microbial inoculants and biological control agents for plant growth and development. This can be used as the development of specific endogenous complexes of crops to promote sustainable agriculture.
Keywords: plant microbiota, plant-microbe interaction, sustainable agriculture, rhizosphere, endophyte, leaf ball
Phytomicrobiome for promoting sustainable agriculture and food security: opportunities, challenges, and solutions
Journal: Microbiological Research
IF: 3.970
Issuing time: 2021.04.05
Corresponding author: Brajesh Kumar Singh
Corresponding Author Unit: Banaras Hindu University, the Holy City of Varanasi, Uttar Pradesh, India
DOI number: 10.1016/j.micres.2021.126763
(source:internet, reference only)
Disclaimer of medicaltrend.org



