ADC linker and latest research progress
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
ADC linker and latest research progress
ADC linker and latest research progress. The linker is not only the part of the molecule that forms a covalent connection between the antibody and the small molecule payload, but also a key element with design properties in targeted drug therapy.
The addition of the linker should not induce aggregation, and it is necessary to ensure acceptable PK characteristics, while limiting the premature release (stability) of the payload in plasma, and enabling the effective release of active molecules at the target site of action. Linkers are divided into two categories: non-cuttable and cuttable.
Non-cleavable linker
ADCs based on non-cleavable linkers must be internalized, and the antibody portion needs to be degraded by lysosomal proteases to release active molecules. Many non-cleavable linkers have been explored during the ADC development process, the most representative one is N-succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylic acid Salt (SMCC), Kadcyla is the type of linker used.
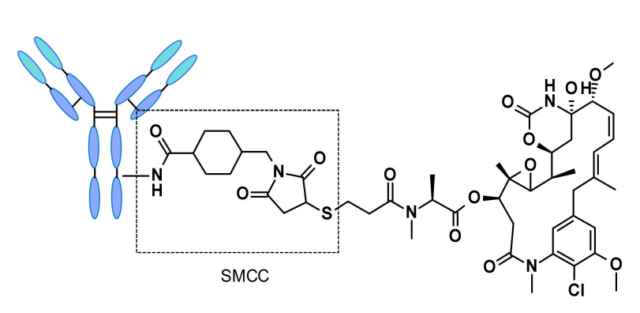
The catabolism of this structure causes Lys-SMC-DM1 to become the main tumor metabolite. In addition, drugs linked to this linker usually cannot exert a bystander effect because the released catabolites are less permeable. Current research mainly focuses on cleavable linkers.
The use of cleavable linkers is equally feasible for both internalized and non-internalized ADC designs, because the release is triggered by the nature of the cleavage site (lysosome and/or tumor environment). Linkers can be divided into two categories: enzyme-dependent and chemical (ie, non-enzyme) dependent.
Chemically dependent linker
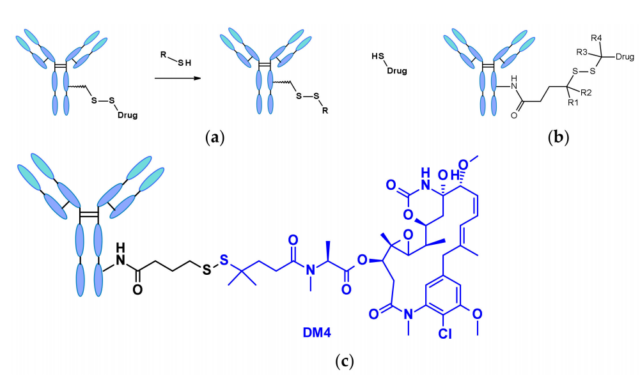
The disulfide bond-containing linker is nucleophilically attacked by thiols to release the active load. Although the reduced form of human serum albumin (HSA) in plasma is the most abundant thiol, it has poor reactivity to macromolecules. The cytoplasm also contains high levels of glutathione (GSH), a tripeptide containing sulfhydryl groups that easily reacts with S-nucleophilic protein. The difference in GSH concentration in blood (micromolar range) and cytoplasm (millimolar range) and the oxidative stress caused by cancer cells contribute to the preferential release of drugs in cancer cells. Linkers containing disulfide bonds are mainly related to maytansinoid-like payloads. The reactivity of disulfide bonds can be adjusted by steric hindrance: α-methyl substitution significantly affects the reduction rate and resistance to thiol-disulfide bond exchange. For example, the linker of SAR-3419 obtains SPDB- by geminal dimethyl substitution. The best anti-tumor activity of DM4.
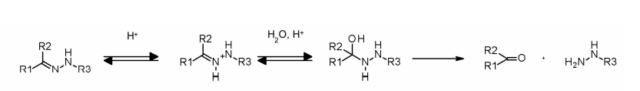
The hydrazone linker shows pH-dependent stability, is stable at neutral pH, and is hydrolyzed in an acidic medium (endosome pH<6, lysosome pH<5) to form corresponding ketones and hydrazines.
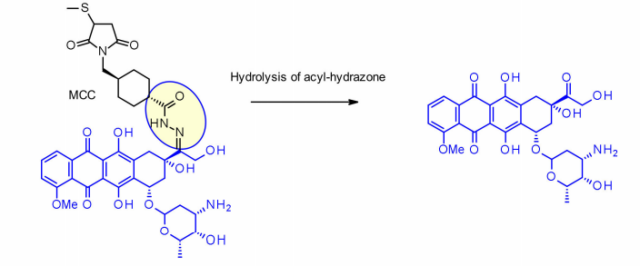
This method has been successfully applied to IMMU-110, which contains a cleavable acylhydrazone linker, which is composed of 4-maleimidomethylcyclohexane-1-carboxylate (MCC) hydrazide and doxorubicin. The keto group is formed during the reaction.
The hydrazone linker is also often used in the payload of the carlimycin family. In this case, the release is triggered by a two-step activation process: the first step is the acid-sensitive hydrazone is hydrolyzed, and the second step is the disulfide bond reduction by GSH , The sulfhydryl intermediate is cyclized. This linker has been used in the marketed Mylotarg and Besponsa, but their stability in plasma is not as expected and not as attractive as other cleavable linkers.
Enzyme-dependent linker
In order to limit the release of the payload before internalization, thereby preventing or minimizing the degradation outside the target cell, the protein component of the lysosome becomes a reasonable place to find enzymes that can degrade ADC and exist in high concentrations.
Cathepsin-B
Cathepsin B is a cysteine protease that exists in the late endosomes and lysosomes of mammals, and is also overexpressed in many cancer cells. Initially, a cleavable dipeptide was used as a cathepsin B substrate as a prodrug of doxorubicin. This work established the dipeptide portion of SAR: the P1 position requires a hydrophilic residue (citrulline or arginine). Acid), while the lipophilic residue at the P2 position enhances plasma stability (phenylalanine, valine or alanine).
In addition, a self-degrading spacer is introduced to promote the entry of enzymes, thereby limiting the steric hindrance of the payload: p-aminobenzyl carbamate (PABA) spontaneously 1,6-eliminates in acidic medium and releases carbon dioxide , P-azaquinone carboxamide and adriamycin. In the end, this discovery transferred from the prodrug to the ADC field, demonstrating the antigen-driven cellular activity of Val-Cit and Phe-Lys dipeptide linkers.
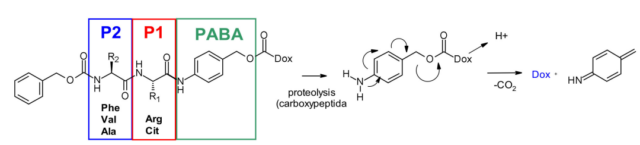
Val-Cit dipeptide is the most commonly used cleavable linker in ADCs. There are currently as many as 25 molecules in the clinical stage, probably because of its overall good plasma stability, release behavior and chemical towability. The two approved ADC drugs (Adcetris and Polivy) both use the same linker mc-VC-PABC, which contains a maleimide spacer, a standard Val Cit dipeptide sequence as a cathepsin substrate, and PABC self-degrading spacer.
Val-Ala dipeptide is also widely used. There are 7 molecules in the clinical stage. The fastest progress is Loncastuximab tesirine, which includes a pegylated spacer to balance the affinity of the payload SG3199, which belongs to the PBD dimer family. Fatty.
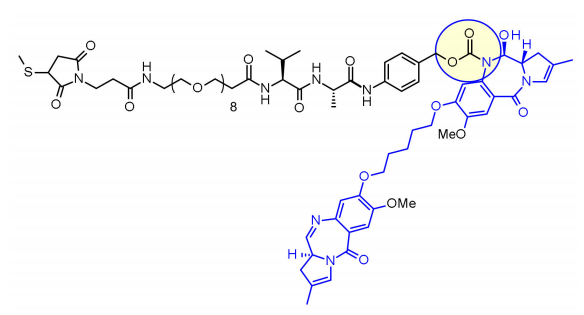
Studies have shown that it is difficult for Val-Cit to achieve high DAR due to precipitation and aggregation. In contrast, the Val-Ala linker allows DAR up to 7.4 with limited aggregation (<10%). Compared with Val-Cit, Val-Ala is less hydrophobic, which explains why this linker excels in lipophilic payloads (such as PBD dimers). The Val-Ala of 7 clinical candidate ADCs The connectors are connected to the PBD.
Some studies have compared Val-Cit and Val-Ala dipeptide structures with MMAE payload linkage. In the case of non-internalized antibodies, both Val-Cit and Val-Ala linkers that bind to engineered cysteine exhibit similar characteristics and perform better than Val-Lys and Val-Arg analogs. performance. In the case of using a random cysteine-conjugated anti-Her2 ADC, Val-Ala shows less aggregation in the high DAR structure compared to Val-Cit. On the other hand, the two linkers showed similar buffer stability, cathepsin B release efficiency, cell activity and histopathological characteristics.
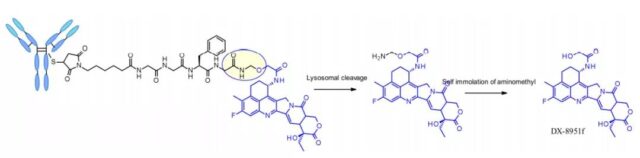
The tetrapeptide Gly-Gly-Phe-Gly shows all the characteristics of a stable and effective cleavable linker, which is used in the ADC drug Enhertu, which is already on the market. Daiichi Sankyo’s Enhertu is a plasma-stable ADC with a DAR of 7.7. It undergoes protease degradation in the lysosome and releases DX-8951f, which is a potent topoisomerase I inhibitor derived from exatecan. Since the linker does not contain a solubilizer, achieving such a high DAR is very impressive because it contradicts the widely established principle that high DAR conjugates may have poor pharmacokinetic characteristics. The self-degrading spacer used here is a simple and compact semi-amination instead of the PABC used in the Val-Cit linker.
Phosphatase and pyrophosphatase
Like cathepsins, pyrophosphatase and phosphatase are also hydrolases that are selectively expressed in lysosomes. In 2016, Merck’s researchers designed a linker containing phosphoric acid and pyrophosphate to match the cathepsin B-sensitive Val-Cit-PABA to deliver glucocorticoids: the phosphate/pyrophosphate part binds to self-degradation Spacer between PABA and payload. After internalization, the payload can be released in the order of cathepsin B, self-degrading spacer and phosphatase (n=1). For pyrophosphate (n=2), another step involving pyrophosphatase may be required.
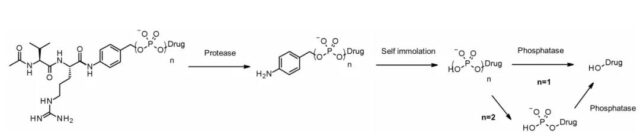
The advantage of this hydrophilic and permanently charged group is solubility. It can not only be bio-coupled with lipophilic glucocorticoid derivatives, but also promote the purification of ADC. The residual linker in ADC is less than 0.10%. ADCs containing phosphoric acid and pyrophosphate are both active in vitro.
The same team of researchers at Merck also developed a unique pyrophosphatase-based linker for releasing hydroxyl-containing payloads dexamethasone and fluticasone propionate.
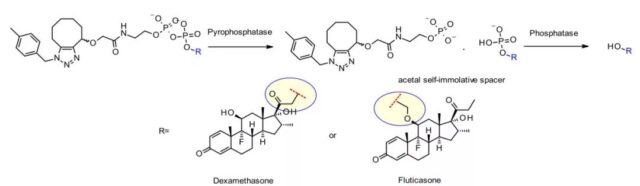
In addition, the nature of the hydroxyl attachment point is critical for effective release. The primary alcohol of dexamethasone works well, while the more hindered secondary alcohol of fluticasone requires an acetal spacer to achieve acceptable release. Both ADCs show good stability in vitro and have strong activity on tumor cell lines.
β-glucuronidase
β-glucuronidase is a type of glycosidase that catalyzes the hydrolysis of β-glucuronic acid residues, and it is highly expressed in lysosomes and tumor stroma. Researchers from Seattle Genetics published a groundbreaking work in 2006. The anti-CD70 ADC uses a linker containing glucuronic acid, which is attached to a self-degrading spacer. This linker exhibits low levels of aggregation, high plasma stability, and strong in vivo efficacy.
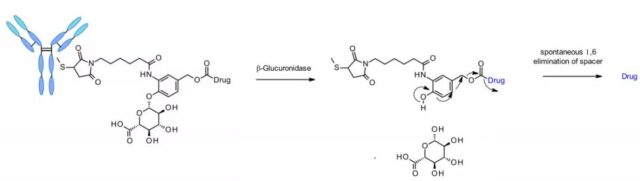
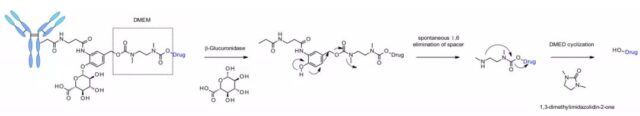
The linker is also applied to other amine-containing payloads, such as camptothecin analogues, SN38, ducamycin, and matrine through an additional dimethylethylenediamine (DMED) self-degrading spacer. The release sequence is from the hydrolysis of β-glucuronic acid to the self-degrading spacer. Another cyclization reaction of DMED occurs spontaneously to form 1,3-dimethylimidazolin-2-one, and finally release the hydroxyl-containing drug. Due to the hydrophilic nature of the linker, this technology makes it easier to prepare ADCs with DAR=8 compared to cathepsin-sensitive linkers.
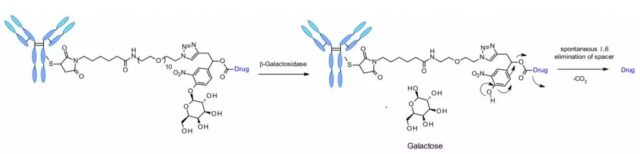
β-galactosidase
Recently, an ADC using β-galactosidase to cleave the linker was reported, which contains a PEG10 spacer. The spacer is replaced by a nitro group to increase the rate of self-degradation. Analogous to the β-glucuronidase linker, its dissociation mechanism involves the hydrolysis of the β-galactosidase moiety, which imparts hydrophilicity to the chemical precursor. Another advantage is that β-galactosidase exists only in lysosomes, while β-glucuronidase is expressed in lysosomes and also in the microenvironment of solid tumors. Studies have shown that in the context of anti-HER2-ADCs releasing MMAE, ADCs containing β-galactosidase linkers are more effective than T-DM1 in vitro and in vivo.
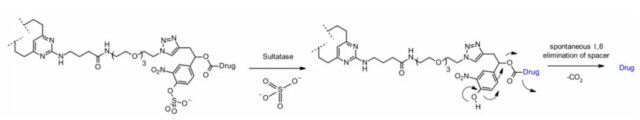
Sulfatase
Recently, linkers cleaved by sulfatase have appeared, and sulfatase is overexpressed in several cancer types, showing potential selectivity. The study involved an anti-Her2 antibody with MMAE as the payload. Compared with the classic cleavable Val-Cit and Val-Ala linkers, the sulfatase linker showed similar efficacy to Her2+ cell lines.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



