2020 New Immunotherapeutic drug technology FDA/EMA approved
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
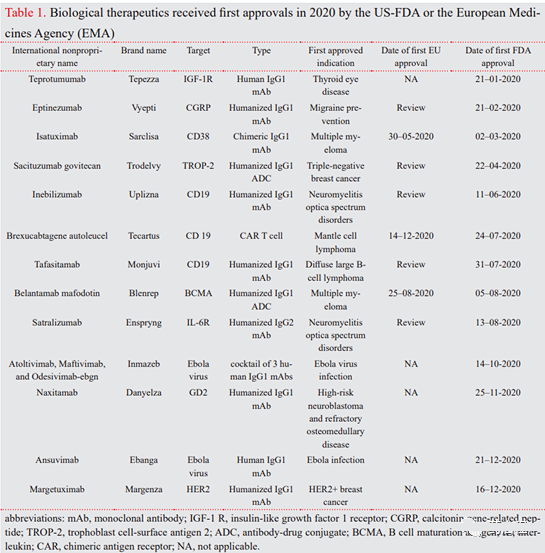
2020 New Immunotherapeutic drug technology FDA/EMA approved. In 2020, among 53 products approved by the U.S. Food and Drug Administration (FDA), there are 12 antibody therapies and one CAR-T cell therapy. Here, this review aims to explain the applications of new immunotherapeutic drugs approved by the FDA/EMA in 2020 (Table 1). They belong to 3 main categories: monoclonal antibodies (mAb), antibody-drug conjugates (ADC) and CAR -T cell therapy.
1. Introduction
For a long time, researchers and pharmaceutical companies have developed a keen interest in immunotherapy that manages and treats a variety of diseases through multiple ways of controlling the immune system (1, 2). In cancer and infectious diseases, immunotherapy is used to activate the immune system to eradicate evading cells, while for autoimmune diseases, transplants, allergies, and wound healing, immunotherapy aims to suppress the immune response to improve safety or tissue regeneration (3, 4).
It can be divided into several categories, including:
a) immunomodulators (for example, cytokines, interleukins, chemokines, and immunomodulatory drugs),
b) monoclonal antibodies,
c) checkpoint blocking,
d) oncolytic viruses ,
E) vaccines,
f) cell therapy, such as chimeric antigen receptor (CAR) T cells (5).
In 2020, among 53 products approved by the U.S. Food and Drug Administration (FDA), there are 12 antibody therapies and one CAR-T cell therapy. Here, this review aims to explain the applications of new immunotherapeutic drugs approved by the FDA/EMA in 2020 (Table 1). They belong to 3 main categories: monoclonal antibodies (mAb), antibody-drug conjugates (ADC) and CAR -T cell therapy.
2. Monoclonal antibodies
In the past few decades, monoclonal antibodies (mAb) or their combined use with other drugs, toxins, and radionuclides have received great attention (7).
There are various techniques (Figure 1). They were first produced in mice using hybridoma technology, which uses the fusion of B lymphocytes from the spleen of immunized animals with an immortal myeloma cell line (8). However, the clinical application of complete murine mAbs is hindered by the production of human anti-mouse antibodies, increased murine mAb clearance, and adverse allergic reactions (9). Therefore, murine mAbs are transformed by chimeric technology, in which the non-human variable domain is combined with the human constant (C) region d. A major advancement in humanized mAb production is through the use of complementary domain-to-determined region (CDR) grafting technology.
In this technique, non-human antibody domains are transferred to human framework sequences while retaining target specificity (10). The technology platform has made progress to obtain fully human monoclonal antibodies. In another technique called phage display, foreign genes are integrated into phage to generate a peptide library on the surface of phage virus particles, thereby screening the phage library to bind the target antigen (11).
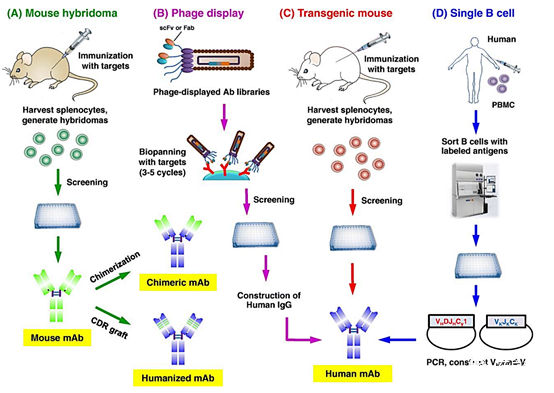
Figure 1. Techniques used to produce therapeutic antibodies: a) Murine hybridomas, including mouse immunization, fusion of harvested spleen cells and myeloma cell lines, and screening to select the desired B lymphocyte clones. The selected clones can be used to produce chimeric or humanized antibodies; b) phage display, including the production of human antibody libraries in phage and selection of single-chain antibodies against the desired antigen that can be used to produce human mAbs; c) immunization with the target antigen Spleen cells are then harvested to produce mouse hybridomas to produce human monoclonal antibodies; d) Single B cell technology, in which peripheral blood mononuclear cells (PBMC) isolated from infected or vaccinated donors are passed Screening by flow cytometry, followed by RNA extraction, RT-PCR, and cell-free protein synthesis (CFPS) to produce human mAb (10).
Transgenic animals with human antibody libraries represent another technique for obtaining fully human therapeutic antibodies after immunization with target antigens. Abgenix and Medarex are two examples of genetically modified mice that produce fully human antibodies for tumor indications (7). However, genetically modified mice cannot accurately mimic the human immune response. Therefore, the single B cell technology has been developed, and its working principle is to sort the B cells labeled with the target antigen from the patient and transform them with Epstein-Barr virus (EBV) to make them immortal (8). In 2020, 10 monoclonal antibodies include 4 humanized antibodies (Nacitalizumab, Tafacitimab, Satralizumab and Inebilizumab) and 3 fully humanized antibodies (antibody mixture of Ansuvimab, Sacituzumab and Atoltivimab, Maftivimab and Odesivimab) And 2 chimeras (Margetuximab) and Isatuximab were approved in the United States or the European Union (EU) (Table 1).
2.1. Eptinezumab in the treatment of paroxysmal migraine
Eptinezumab (VyeptiTM, Lundbeck) is a humanized IgG1 mAb that blocks calcitonin gene-related peptide (CGRP) α and β. It is produced in Pichia pastoris cells through recombinant DNA technology (12). Vyepti TM was approved as the first intravenous (IV) migraine prophylaxis in the United States in February 2020. The FDA approval was mainly based on positive data from two clinical trials (Trial 1/NCT02559895 and Trial 2/NCT02974153), which involved 1741 subjects with chronic or incidental migraine (13). The recommended dose is 100 mg every 3 months, although some patients may benefit from a single dose of 300 mg (14).
2.2. Nacitalizumab in the treatment of high-risk neuroblastoma
Naxitamab (DanyelzaTM, Y-mAbs Thera peutics) is a humanized IgG1 mAb targeting GD2 (hu3F8), and GD2 (hu3F8) is a disialylganglioside highly expressed in neuroblastoma. Its binding to GD2 stimulates both complement-dependent cytotoxicity (CDC) and antibody-dependent cell-mediated cytotoxicity (ADCC) (15). Naxitamab is produced in the Chinese Hamster Ovary (CHO) cell line.
The product was first developed at the Memorial Sloan Kettering Cancer Center (MSK) in New York, and then the commercial rights were fully licensed to Y-mAbs Therapeutics for the treatment of neuroblastoma, osteosarcoma and other GD2-positive cancers (16).
Nalcitimab combined with granulocyte macrophage colony stimulating factor (GM-CSF) was approved by the U.S. FDA in November 2020 for the treatment of patients with relapsed or refractory bone or intraosseous high-risk neuroblastoma (one year old) Above) bone marrow (17).
The drug approval is based on the results of two open-label trials (Trial 1/NCT03363373 and Trial 2/NCT01757626) in 97 patients with high-risk neuroblastoma of the bone or bone marrow. The subjects received an intravenous infusion of nalcitizumab (3 mg/kg) on days 1, 3, and 5 of each 4-week cycle, and subcutaneous injection of GM- at a dose of 250 µg/m2/day within a few days CSF-4 to 0 and 500 µg/m2/day, day 1 to 5 (18). The results showed that the overall response rates of Test 1 and Test 2 were 45% and 34%, respectively.
2.3. Application of Tafasitatamab and Isatuximab in blood cancer
Tafasitatamab (MonjuviTM, MorphoSys Inc.) is a humanized IgG1 anti-CD19 mAb with a modified Fc domain that leads to increased Fcγ receptor affinity (19). It is produced by recombinant DNA technology in CHO. In July 2020, Monjuvi TM combined with lenalidomide was approved by the FDA for the treatment of adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) not suitable for autologous stem cell transplantation (20). The efficacy of tafasitamab was evaluated in a phase 2 study (NCT02399085) in 81 patients with recurrent lymphoma. Participants received intravenous tafasitatamab (12 mg/kg) combined with lenalidomide for a maximum of 12 cycles. The results showed that the best overall response rate was 55%, the complete response rate was 37%, and the partial response rate was 18% (21).
Isatuximab (SarclisaTM, Sanofi) is a chimeric IgG1 monoclonal antibody produced in CHO cells using recombinant DNA technology to target the CD38 receptor overexpressed in plasma cells of patients with multiple myeloma.
In March 2020, the US FDA approved intravenous etuximab combined with pomalidomide and dexamethasone for the treatment of adult patients with multiple myeloma. These patients have received at least two treatments in the past, including lenalidomide and dexamethasone. Bortezomib (22). Isatuximab also received a positive evaluation for the same indication in the EMA in May 2020 (19). The approval of Sarclisa TM is based on the results of a multinational, randomized, open-label Phase 3 study (ICARIA-MM, NCT02990338) of 307 patients with relapsed and refractory multiple myeloma. The results showed that the median progression-free survival of the isatuximab-pomalidomide group increased from 6.47 months to 11.53 months compared with the group receiving pomalidomide and dexamethasone (23).
2.4. Margetuximab in the treatment of HER-2 positive breast cancer
Margetuximab (MargenzaTM, MacroGen ics) is a chimeric IgG monoclonal antibody targeting the HER2 receptor of breast cancer. Margetuximab binds to the same epitope as trastuzumab, but its Fc domain has been modified to enhance binding to activating FcγRIIIA (CD16A) and reduce its affinity for inhibitory FcγRIIB (CD32B) receptors (24). The FDA approved margetuximab combined with chemotherapy for the treatment of adult patients with HER2-positive breast cancer who have previously received at least two anti-HER2 therapies (25). The efficacy of margetuximab was evaluated in phase 3 SOPHIA (NCT02492711) recruiting 53 patients with HER2+ metastatic breast cancer. The results showed that the median progression-free survival of the margetuximab treatment group was 5.8 months, while that of the trastuzumab control group was 4.9 months. In addition, compared with trastuzumab, margetuximab reduces the risk of disease progression by 24% (26).
2.5. Teprotumumab in the treatment of thyroid ophthalmopathy
Teprotumumab (TeppezaTM, Horizon Therapeutics) is a humanized IgG1 antibody against insulin-like growth factor-I receptor (IGF-IR). It is produced in CHO-DG44 cells. Teppeza TM was designed by Roche Ltd. to treat cancer, including breast cancer, non-small cell lung cancer, solid tumors, Hodgkin’s disease, non-Hodgkin’s disease and sarcoma (27). In January 2020, the FDA approved Tepezza TM as the first drug for the treatment of thyroid ophthalmopathy, which is an autoimmune inflammatory disease characterized by more than one eyelid retraction, eyelid retardation, conjunctivitis and bulging eyes (19) .
TepezzaTMapproval is based on positive data from two clinical trials (Phase 2, NCT01868997 and Phase 3, NCT03298867), which consisted of 170 patients with active thyroid eye disease. Participants received teprotumumab infusion (10 mg/kg on day 1, and 20 mg/kg every three weeks for the remaining 7 infusions. The results showed that 71% of the drug treatment group in the phase 2 study and 83 in the phase 3 clinical trial % In contrast, 20% and 10% of those who received placebo had a reduction of more than 2 mm in exophthalmos (protrusion) (28).
2.6. Application of Satralizumab and Inebilizumab in autoimmune CNS diseases
Satralizumab (EnspryngTM, Genentech Inc.) is a humanized IgG2k anti-interleukin 6 (IL 6) receptor monoclonal recirculating antibody, produced by recombinant DNA technology in the CHO cell line (29). The antibody was mainly developed at home and abroad and was licensed to Roche in 2016 (19).
In August 2020, satralizumab was approved by the FDA as the first subcutaneous treatment for neuromyelitis optica spectrum disorder (NMOSD) or Devic disease in adult patients with positive anti-aquaporin 4 (AQP4) water channel autoantibodies.
NMOSD is a rare autoimmune disease of the central nervous system characterized by injury and acute inflammation of the optic nerve and spinal cord (30). The safety and effectiveness of satralizumab were studied through two 96-week clinical studies: SAkuraStar (NCT02073279) and SakuraSky (NCT02028884), including 95 (64 anti-AQP4 antibody positive) and 76 adult patients (52) Anti-AQP4 positive), respectively (31).
The results showed that compared with 41.1% in the placebo group, the number of NMOSD recurrences was reduced by 76.5%. In addition, 91.1% of satralizumab-treated patients did not relapse after 96 weeks, compared with 56.8% in the placebo group (21).
Inebilizumab (UpliznaTM, Viela Bio) is a humanized anti-CD19 IgG1 mAb for the treatment of autoimmune diseases associated with CD19-expressing B cells. It is produced by recombinant DNA technology in CHO. Inebilizumab depletes CD19+ B cells and plasmoblasts, which are responsible for the production of autoantibodies against AQP4.
In June 2020, inebilizumab was approved by the FDA for the treatment of NMOSD in AQP4-IgG-positive adult patients. The drug is still undergoing clinical evaluation for kidney transplant desensitization, myasthenia gravis, and IgG4-related diseases (32).
The FDA approval of Uplizna TM is based on the positive results of a 28-week Phase 2/3 study (NCT02200770) consisting of 230 adult participants with and without AQP4-IgG antibodies. Subjects received two doses of 300 mg Inebilizumab as monotherapy or placebo on Day 1 and Day 15. Among patients with positive anti-AQP4 antibodies, the risk of NMOSD recurrence was reduced by 89%, compared with 58% in the placebo group (19).
2.7. The role of Inmazeb TM and Ansuvimab in Ebola virus infection
Atoltivimab, maftivimab and odesivimab (InmazebTM, Regeneron) are a combination of three fully human monoclonal IgG1 antibodies-atolt ivimab (REGN3470), maftivimab (REGN3479) and odesivimab (REGN3471)-they bind to EBOV) responsible for cell input . InmazebTM is produced by recombinant DNA technology in CHO using VelociSuite® technology in VelocIm mune® mice immunized with coding DNA or purified recombinant EBOV glycoprotein.
This antibody cocktail was approved by the FDA in October 2020 and became the first drug to treat infections caused by EBOV in Zaire in children and adults (33). In the PALM study (NCT03719586), the effectiveness of Inmazeb was evaluated for 28 days, of which 154 enrolled patients received a single intravenous infusion of 50 mg of each mAb, and 168 patients received remdesivir as a control . During the intervention period, 33.8% of Inmazeb-treated patients died, compared with 51% of participants receiving the control drug (34).
Ansuvimab (EbangaTM, Ridgeback Biotherapys) is a human IgG1 monoclonal antibody that binds to the glycoprotein on Zaire’s EBOV to prevent viruses from entering cells. Ansuvimab is produced in CHO cells through recombinant DNA technology. In December 2020, Ebanga TM was approved by the FDA for the treatment of Zairian EBOV infection in adults, children or newborns born to mothers infected with Ebola virus (35).
The safety and effectiveness of EbangaTM were evaluated in a clinical trial (Trial 1/NCT NCT03719586) in 342 Zairian patients with EBOV infection. 173 subjects received a single intravenous infusion dose of 50 mg/kg EbangaTM. Of the 174 participants treated with EbangaTM, 35.1% died, while 49.4% of 168 patients who received remdesivir as a control died (36).
3. Antibody conjugated drugs
Antibody-drug conjugates (ADC) are designed to specifically deliver anti-tumor drugs to target antigens by coupling with antibodies (37). The design of ADC depends on the appropriate selection of four key components: target antigen, antibody construct, cytotoxic agent and linker (Figure 2) (38). Early ADC development used murine mAbs, but due to their immunogenicity, Low efficacy, suboptimal targeting, and insufficient tumor selectivity compared with normal tissues have not produced satisfactory results in clinical trials (39). To solve these problems, murine antibodies have been replaced by humanized or fully human antibodies.
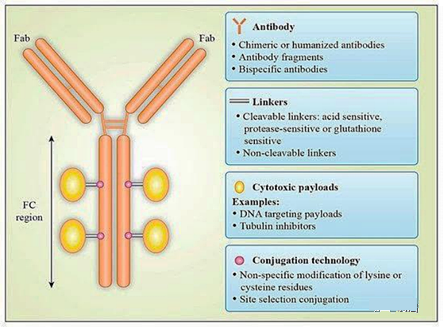
Figure 2. Schematic diagram of the structure of the antibody-drug conjugate: The antibody-drug conjugate (ADC) consists of three main building blocks: antibody, cytotoxic payload, and a linker that binds these two parts. Each part should be optimized to improve the potency, safety and efficacy of ADC (44)
Due to the low efficacy of the payload and the instability of the ADC in the blood circulation leading to systemic toxicity, the first-generation ADC composed of mAb linked to conventional chemotherapeutic drugs has not been successful (40). The second-generation ADC uses effective cytotoxic agents, targeting tubulin or DNA, with IC50 values in the sub-nanomolar range. However, their use may be affected due to the heterogeneity of the final conjugate, the limited penetration of solid tumors, the development of drug resistance, and the narrow therapeutic index (41).
Another problem is that the linker chemistry, as a key part of ADCs, determines their stability and toxicity in the blood circulation. Linkers usually react with lysine side chains or sulfhydryl groups in the hinge region of antibodies, resulting in a heterogeneous ADC population with an average drug-to-antibody ratio (DAR) of 3-4 (39). After the antibody binds to the target antigen, the payload can be released under extracellular conditions; otherwise, due to the proteolytic degradation of the entire ADC molecule or the cleavage of the linker at low pH or degradation mediated by the proteasome, the ADC is internalized And release the payload (38).
In the third-generation ADC, multiple technologies such as THIOMAB, ThioBridge, SMARTag, and SMAC-Tag are used, with special attention to site-specific conjugation to ensure homogeneous ADCs (42). In addition, in order to overcome drug resistance, people are paying attention to new payloads with broad-spectrum activity against non-proliferating cancer cells, such as topoisomerase inhibitors, pyrrolobenzodiazepine derivatives of tricyclic antibiotics, and duocarmy cins. MylotargTM (gemtuzumab ozogamicin) is the first ADC approved by the US FDA for CD33-positive acute myeloid leukemia (43).
To date, there are eight different ADCs, including Gemtuzumab ozogamicin (MylotargTM), Brentuximab vedotin (AdcetrisTM), Trastuzumab emtansine (KadcylaTM), Inotuzumab ozogamicin (BesponsaTM), Polatuzumab vedotxin (Poliutuzumab suedodotTM), Polatuzumab vedotxin (Enolivyb vedotTM) It has been approved by the US FDA, and dozens of drugs are in the preclinical and clinical development stages (37). In 2020, TrodelvyTM and BlenrepTM have been approved by the US FDA for the treatment of cancer.
3.1. Sacituzumab govitecan in the treatment of metastatic breast cancer
Sacituzumab govitecan (TrodelvyTM, Immunomedics Inc.) is a third-generation ADC that contains a humanized IgG1 anti-Trop-2 antibody that is linked to SN-38 as a topoisomerase I inhibitor. SN-38 is covalently attached via a hydrolyzable CL2A linker, which contains a short PEG7 chain for improved solubility (Figure 3).
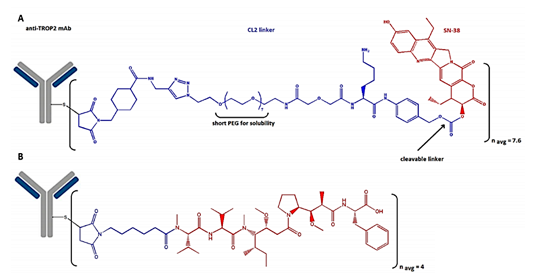
Figure 3. A) The structure of sacituzumab govitecan, which consists of an anti-Trop-2 antibody connected to SN-38 via a hydrolyzable CL2A linker containing a short PEG7 chain, and B) belantamab mafodotin is connected to a targeted B cell maturation antigen (BCMA) The antibody composition of is connected to the microtubule inhibitor monomethyl auristatin F (MMAF) via an uncleavable bond (50).
After binding to the TROP2 receptor, Trodelvy TM is internalized and subsequently releases SN-38, leading to DNA damage and cell death (45). In April 2020, sacituzumab govitecan received accelerated approval from the FDA for the treatment of metastatic triple-negative breast cancer in adult patients who had previously received at least two therapies (46). The effectiveness and safety of TrodelvyTM were demonstrated in the multi-center, single-arm clinical trial IMMU 132-01 (NCT 01631552), which enrolled 108 patients with metastatic triple-negative breast cancer.
Saci tuzumab govitecan was administered intravenously at 10 mg/kg every 21 days on days 1 and 8. Based on the results, the estimated overall response rate was 33.3%, and the median duration of response was 7.7 months (47). Sacituzumab govitecan is undergoing phase III clinical trials for breast cancer and phase II studies for urothelial cancer in the United States and the European Union. It is also studying brain metastases, glioblastoma, endometrial cancer, and prostate cancer (45).
3.2. Belantamab mafodotin in the treatment of multiple myeloma
Belantamab mafodotin (BlenrepTM, Glax oSmithKline) contains a humanized IgG1 antibody targeting B cell maturation antigen (BCMA), which is linked to the microtubule inhibitor monomethyl auristatin F (MMAF) via an uncleavable bond (Figure 3) (48).
The drug was developed using BioWa’s licensed POTELLIGENT technology to improve the efficacy of ADC by reducing the fucose content in the carbohydrate structure of the mAb produced by the fucosyltransferase knockout CHO cell line. After BlenrepTM binds to BCMA and internalizes the cell, the linker is cleaved by lysosomal proteases to provide intracellular delivery of MMAF, followed by cell cycle arrest.
In August 2020, the FDA and EMA approved Belantamab mafo dotin for the treatment of adult patients with relapsed or refractory multiple myeloma. These patients have previously received at least four treatments, including anti-CD38 mAb, proteasome inhibitor, and immunomodulation(19).
Belantamab mafo dotin was evaluated in the open-label, multicenter DREAMM-2 (NCT03525678) study. Patients receiving a 2.5 mg/kg dose every 3 weeks showed an overall response rate of 31%, while 73% of responders showed a response duration of more than 6 months (49).
4. CAR-T cell therapy
CAR-T cells are composed of T cells isolated from peripheral blood and CAR (Chimeric Antigen Receptor) (51). For CAR T cell therapy, T cells are separated from the patient’s blood, transfected with plasmid DNA or transduced with viral vectors, and new antigens are presented on the surface of T cells, thereby redirecting T cells to specific antigens in tumor cells ( 52).
The CAR molecule is composed of three parts: 1) An extracellular domain containing a single-chain fragment variable for a specific target antigen. This domain is connected to 2) the transmembrane domain, a part of CD3, CD8, CD28 or FcεRI, which binds to 3) the intracellular domain, with or without activation domain (CD28, CD27, CD134, CDB7 or CD3ζ) The second costimulatory factor (CD28 or 4-1BB) (52). The structure of different CAR generations is characterized by different intracellular signal domains (Figure 4). The original CAR framework contained an intracellular CD3ζ signal domain, which had insufficient signal capacity.

Figure 4. A) Generation of chimeric antigen receptor (CAR). The first generation CAR contains an intracellular CD3ζ signal domain, if combined with the costimulatory domain (mainly CD28), the second generation will be formed. The third-generation CAR involves additional costimulatory domains, such as CD28 or CD137. The fourth-generation CAR contains additional gene cassettes expressing cytokines (51). B) Anti-tumor cytotoxicity mechanism of CAR T cells: T cells are activated by phosphorylation of immunoreceptor tyrosine activation motif (ITAM), and then enhance cytokine secretion (including IL-2, IL-4, IFN-γ) , IL-12 and TNF) and T cell proliferation. IL-12 enhances the function of immune cells, such as NK cells and macrophages. Activated CAR-T cells mainly kill tumor cells by secreting perforin and granzyme particles and the Fas/Fas-L death receptor pathway (55).
The first-generation CARs can specifically detect tumor antigens, but due to their reduced proliferation capacity, their therapeutic effects in vivo are not ideal. The second generation CAR also contains costimulatory domains, such as CD28 and 4-1BB, which show improved cell proliferation. In the third-generation CAR, two different costimulatory signals such as CD28 and CD137 participate simultaneously. The fourth-generation CAR contains cytokines and improves tumor cytotoxicity by overcoming the immunosuppressive tumor microenvironment (51).
The cytotoxicity of CAR-T lymphocytes is based on two main pathways: (1) the secretion of perforin and granzyme particles; (2) activation of death receptor signals through Fas or TNF receptors. Because CAR specifically binds to tumor-associated antigen (TAA) on cancer cells, T cells are activated by phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM), which triggers cytokine secretion and T cell proliferation And cytotoxicity (53).
KimriaTM and YeskartaTM are CAR T cell drugs approved by the US FDA in 2017 for the treatment of leukemia and lymphoma (54). In July 2020, the FDA approved the third CAR T cell drug Tacartus TM for the treatment of mantle cell lymphoma (MCL).
Brexucabtagene autoleucel (TecartusTM, Kite Pharma) is an anti-CD19-transduced CD3+ cell designed for autologous T cell immunotherapy in adult patients with relapsed or refractory mantle cell lymphoma (MCL). TecartusTM contains patient T cells, which are genetically reprogrammed by inserting artificial receptor genes using MSCV-based γ-retroviral vectors to express an anti-CD19 single-chain variable fragment (scFv) that is co-stimulated with CD28 CAR domain and CD3-zeta signal domain. The patient received a modified T cell suspension via intravenous infusion.
The modified T cell with CAR on its surface can bind to the CD19 protein on the cancer cell membrane, and then the CD28 costimulatory domain and the CD3-zeta signal domain initiate the downstream signal cascade, which leads to T cell activation and secretion of inflammatory cytokines and chemosin (56). This series of events leads to the death of target cells. Each dose of TecartusTM contains 2×106-2×108 CAR-positive live T cells per kilogram of patient body weight, with a volume of approximately 68 ml (57).
Based on a multi-center, single-arm trial ZUMA-2 (NCT02601313), the FDA accelerated the approval of TecartusTM in July 2020. The trial recruited 74 people who had previously received anthracyclines or bendamustine and Bruton tyrosine kinase Inhibitor chemotherapy for patients with relapsed or refractory MCL. The results showed that the complete remission rate was 62%, and the effective rate was 87% (58)
5. Conclusion
Immunotherapy is a rapidly developing field. In 2020, the FDA/EMA approved a series of new treatment methods. In addition to the therapeutic indications for cancer, mAbs were approved for the first time this year for the treatment of EBOV infection, migrane, an autoimmune central nervous system disease and thyroid eye disease.
This year also brought two ADCs for the targeted treatment of metastatic breast cancer and multiple myeloma with monoclonal antibodies. In addition, currently five CAR-T cell therapies have been approved by the FDA for marketing. Despite the complexity and ambiguity of technological development, ADC and CAR-T cell therapy have great potential in future cancer treatments.
~~~~~End~~~~
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



