Data in the development of artificial intelligence drugs
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Data in the development of artificial intelligence drugs
Data in the development of artificial intelligence drugs. The role and development trend of data in the research and development of artificial intelligence drugs.
Drug molecular design is the sway of thinking in drug discovery. Let us taste the works of drug molecular designers together. It takes time for finished drugs to be marketed. Original design is wonderful.
In recent years, “Artificial Intelligence” (AI) has had a profound impact in image recognition and other fields, but in the field of drug discovery, this application still has limitations, one of the reasons is the existence of cognition of the data it uses problem. Today, let’s discuss artificial intelligence in different fields (such as image, speech, chemistry and biology) data, and learn from its experience to improve the knowledge of drug discovery data, in order to further gain on the basis of understanding of biological systems. Sufficient amount of high-quality data, exploring new ways of drug research and development, and promoting the development of artificial intelligence in the field of drug discovery.
When it comes to artificial intelligence, the image and voice recognition fields (such as automatic passport control and “virtual assistants”) that may first attract public attention. From a technical point of view, a possible starting point for the development of image recognition is Schmidhuber and his colleagues published in Neural Comput in 2010 on the recognition of handwritten characters.
With the emergence of the AlexNet paper published by NIPS in 2012, artificial intelligence began to emerge in the field of handwritten character recognition, and successfully used deep neural network methods for image classification.
This breakthrough does not only come from the design of the algorithm level (such as the use of continuous convolution (Successive Convolution), pooling layer (Pooling Layers), rectified linear (ReLU) unit, data augmentation (Data Augmentation) and data loss layer ( Dropout Layers)), also benefit from the use of a large amount of marked image data and high-speed GPU.
However, can the successful algorithms and data processing methods in the image field be directly used in the field of drug development? The answer is obviously not suitable, let’s look at the possible reasons.
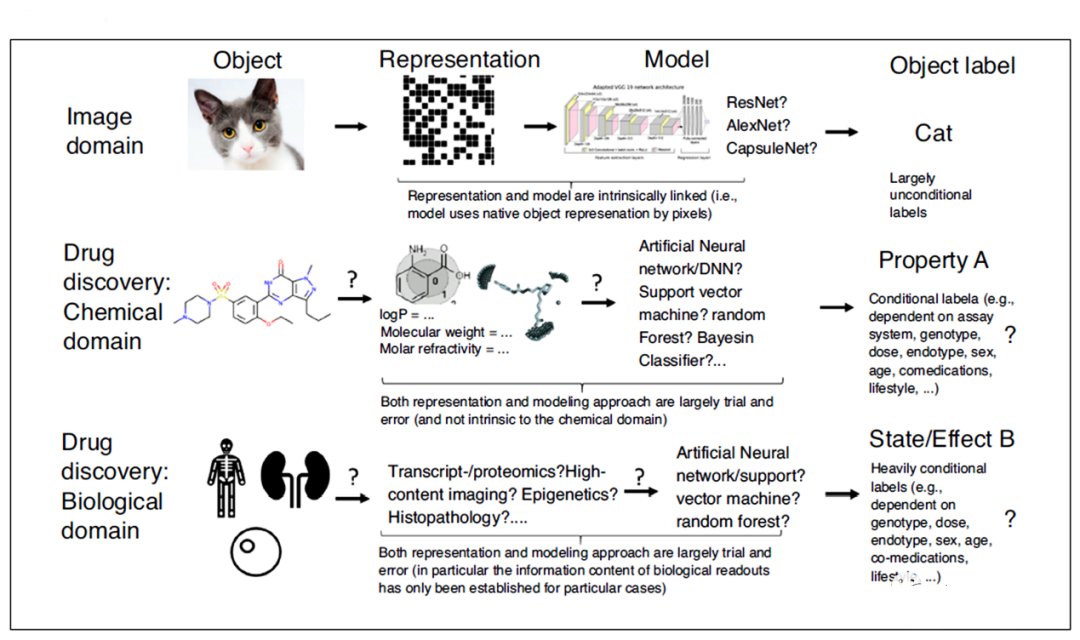
1. Representation of chemical data
In the field of deep learning, data used for learning tasks, especially the amount of data, distribution characteristics and inherent deviations are very important. Unlike image data, autonomous driving, and astronomy, which are often more than 10^9, the amount of effective data in chemistry and biology is relatively small (see “Development thinking and future prospects of artificial intelligence in drug development”).
In addition, the acceptable representation methods, data labeling methods and data mining potential of data in different fields are also very different. In the image field, the research target is usually represented by pixels, and these pixels have a spatial arrangement with each other. Therefore, as long as the shape or color on different pixel values is used as input, the algorithm can be visually distinguished (the same as humans).
For images, although model selection (algorithm structure and hyperparameter selection) also needs to be considered, in most cases of image recognition, pixels are directly used as input. Similarly, the output in image recognition can usually be image-based content (no need to consider the external environment). In contrast, in the fields of chemistry and biology, how to display information to a computer is very subtle.
For example, in the field of chemistry, about 3000 descriptors used to describe chemical molecules have been reported. We still don’t know which descriptor can capture the most critical molecular properties, or we have not yet established the most systematic descriptors. Some molecular characteristics are defined by local characteristics (for example, hydrogen bonds or electric charges), some molecular characteristics are defined by the distribution of characteristics on the molecular surface (for example, lipophilicity), and some molecular characteristics depend on the external environment (for example, The combination of bodies depends on the complex balance of enthalpy and entropy factors).
Therefore, when characterizing chemical data, people can often choose to represent molecules as graphs, curved surfaces, biological activity diagrams, or use other physical and chemical properties. There are many forms, but in a given situation, which molecular properties should be considered. It is predicted that the current research is still limited. In other words, we cannot determine which molecular property is the most important for a given prediction target.
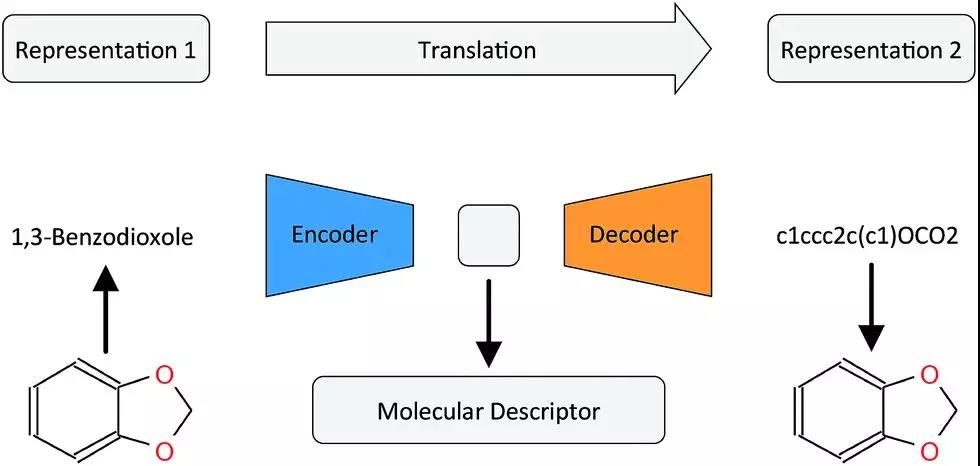
Recently, a lot of work has been used to study how to better represent chemical molecules. Researches believe that the molecular structure representation method used to display connection information and the representation method used for model generation have completely different goals. The molecular connection table (shown graphically or in SMILES format) is used to indicate which bonds exist between atoms in a molecule.
However, considering that the molecule is actually a dynamic object existing in a 3D state (conformation, tautomerism, etc.), this way of expression cannot capture the attributes related to model generation in all situations.
For example, the link table/SMILES format cannot capture the inversion of the boat-chair conformation such as cyclohexane; it also cannot express more complex stereochemical properties (such as axial chirality); it can only represent two electronic bonds; it cannot capture hydrogen. The directionality of the bond makes it impossible to distinguish between intramolecular and intermolecular interactions that affect solubility.
Therefore, it is necessary to distinguish between the information used to indicate the connection of molecules and the representation used for model generation. For the latter, characteristics such as pharmacological points related to receptor interactions should be considered first. However, the data generated for this type of model is scarce. Therefore, for a long time in the future, finding the appropriate molecular representation will always be the focus of artificial intelligence in the field of drug development.
In recent years, there have been some intersections between data representing structural information and model-generated data. For example, the SMILES representation method using molecular structure has achieved a certain breakthrough in the prediction of the result of chemical reactions. In some articles, this type of descriptor is superior to other types of descriptors in terms of performance prediction.
In most cases, we don’t understand which molecular features are really important.
First of all, it is not easy to input a molecule for learning into the computer, because the representation of the molecule depends on the goal of the built model;
Secondly, the choice of machine learning model is subjective. Assuming that molecular features are related to the basic functional form of molecular attributes, whether the features are additive or function in other ways, it is necessary to determine this relationship from the data based on experience. A large amount of data, but due to the size of the chemical space and the current limited amount of data, the available data is still very small;
Finally, the labeling of chemical molecules largely depends on the biological environment (such as whether the nitrogen has been protonated, the dielectric constant of the binding site, the method of measuring molecular solubility, etc.).
Therefore, in principle, we cannot yet clearly label our chemical data in the context of biological systems.
2. Representation of biological data
Similar considerations apply to biological data modeling, and there may even be more problems. It is difficult for us to judge which “descriptor” of the biological system is related to the immediate target problem, even on the label that the descriptor may use:
For example, how to define a disease based on its cause, mechanism, or level of symptoms (where the levels are often different from each other, and some individuals with a given genetic background show very different symptoms, or none at all)? Another example is what is drug interaction? How do we define it? Regarding its relationship to the dose of the drug used, what is the effect of the individual’s genotype (certain drug-drug interactions are only observed in some individuals), and how we deal with the relationship between frequency and event severity in the description ? Just like chemical data, biomarkers are also unclear and are largely determined by specific circumstances. Given a specific test format and parameters, we may be able to specify cytotoxicity, but whether the compound causes hepatotoxicity in the organism depends on the tested species (and strain), the route of administration, the dose, the measurement endpoint considered, and many Other factors (age, gender, co-medication, etc.).
Even so, the control animals showed a certain degree of liver damage in histopathology: is it more meaningful to observe toxicity frequently or infrequently, but the observed toxicity is generally more toxic? In addition, contrary to labels in images, biological systems are not static and will evolve over time, but there is no precedent in the image field that can be used for reference.

When labeling in the biological field, another issue that needs to be distinguished is the difference between “controlled” and “uncontrolled” data. For example, the former is an accurate measurement from a precise device, and the latter is subjectively determined by the image annotation selected by the histopathologist. In principle, uncontrolled variability will bring noisy data, which will reduce the performance of the model.
However, in fact, this is not a black-and-white problem. Even the controlled data part will cause uncontrolled errors in actual operation due to different operating methods/personnel. It can be seen that due to the extremely conditional and multidimensional nature of biotags, assigning tags is not an easy task, and machine learning algorithms need to learn from the labeled data, which leads to the application of artificial intelligence to drug discovery. There is a question of how to mark.
Therefore, we can conclude that areas where artificial intelligence has been successful, such as image classification, are different from the data available in chemistry and biology in the field of drug discovery in the following ways:
(i) The amount of data available;
(ii) The ability to show it to the computer in a suitable form;
(iii) Machine learning algorithms that are essentially consistent with the available data;
(iv) The possibility of assigning valid tags to data. In short, there is a difference between identifying objects on images and identifying safe and effective drugs. It is represented by the field of biology, where systems usually do not follow clearly defined rules.
On the contrary, the definition of biological system is based on different biological levels (such as transcriptomics, proteomics, and metabolomics levels, but also considers the time and space resolution of intracellular and intercellular signal transduction from the cell to the biological level. The level of epigenetic and functional interaction). In addition, observations in the field of biology are highly constrained (depending on a large number of parameters), and this constraint is usually unknown.
3. the development direction of artificial intelligence in drug discovery
The previous discussion briefly explained why the current application of artificial intelligence in drug discovery is not simple. This is largely because it is difficult to generate and label chemical, biological, and physiological data related to efficacy and safety. Currently, what we often do is “build models wherever the data is”; but there are still few real breakthroughs in the field.
Just having “data” does not help (to some extent, it may be useful in other fields, but not in drug discovery): AI data used for drug discovery must be in the correct format, available in a format, and used for the correct purpose In order to bring real changes to the field of drug discovery.
Having said that, people have realized that drug discovery data needs to be better organized and managed than traditional methods. As the overall trend develops, we can better perform data search, classification (when possible) and nearest neighbor search, which are also effective.
Nevertheless, if artificial intelligence is to enter a new level in drug research and development, it needs to go beyond the limitations of current data, and based on their existing existing information, determine what data we need to solve the problem with in vivo safety and effectiveness. Related issues.
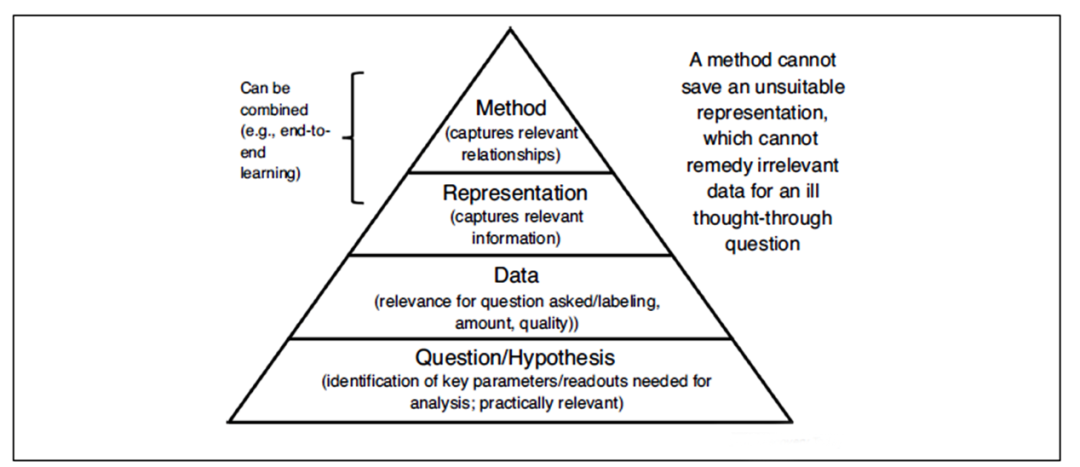
Scientific questions and assumptions are the starting point of any model. We have a hypothesis, for example, that the overexpression of a certain gene is related to a certain disease. Then, this assumption allows us to obtain data in a targeted manner, express it in an appropriate way, and finally analyze the data using appropriate methods. However, the current data often used for drug discovery is not the case.
On the contrary, data is often generated in a non-hypothetical manner (as long as the technical means is suitable for large-scale measurement), and then the parameters are used for subsequent “forced patchwork exercises”. Although it is easier to mine the data, it is not clear that the data obtained in this way is compared with Whether the real situation in the body environment is relevant.
In order to be able to truly use chemical and biological data for drug discovery, we need to go beyond the data “pushing” generated by pure measurement technology to “pull” the generation of data to meet the needs of scientific assumptions.
The data available in the field of drug discovery is essentially different from other areas where artificial intelligence has made great progress (such as image and speed recognition). To some extent, this is related to the difficulty in defining the relevance of specific endpoints (obtaining valid labels for the success of machine learning models); part of the reason is the lack of understanding of specific biological systems.
In many cases, it is difficult to label life science data (due to biological differences, markers are dependent on precise measurement settings, interdependence between markers and the environment, and inconsistent naming methods, etc.), this is useful for artificial intelligence in drugs In terms of discovery, it is a serious problem.
To really advance the development of this field and move from the application in ligand discovery to the application in drug discovery, we need to understand why data is generated and what data needs to be generated, and this first requires a better understanding of biology.
Only when we can measure and capture the relevant biological endpoints in vivo can we make greater progress in this field and apply the currently available computational algorithms to clinical compound efficacy and safety in the field of drug development. the study.
(source:internet, reference only)
Disclaimer of medicaltrend.org



