Peptide drug development strategy targeting protein-protein interaction
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Peptide drug development strategy targeting protein-protein interaction
Peptide drug development strategy targeting protein-protein interaction. Protein-Protein Interaction (PPI) participates in the regulation of many important cellular processes in the organism, such as cell proliferation, division, apoptosis, necrosis, and protein transcription and translation.
There are approximately 130,000 PPIs in the human body, many of which are closely related to the occurrence and development of major diseases such as cardiovascular disease and cancer. The development of PPI-type targets and the development of their agonists or inhibitors have always been the frontiers in the field of international innovative drug research and development.
At present, the PPI drugs that have been marketed mainly include cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors (such as palbociclib, ribociclib, and abemaciclib), checkpoint inhibitors (such as nivolumab and Atezolizumab) and trastuzumab targeting HER2 protein, these drug molecules are mainly divided into antibody and chemical small molecule drugs.
Antibody molecules are difficult to reach intracellular targets due to their low cell permeability, and the range of drugs that can be developed is limited. On the contrary, small molecule drugs (<500 Da) have always been the preferred molecular category for the development of intracellular target drugs due to their good stability, oral bioavailability and transmembrane ability.
For example, the small molecule drug navitoclax that targets PPI can enter cells and bind tightly to the anti-apoptotic protein BCL-2, blocking the interaction between BCL-2 and the pro-apoptotic protein BAK/BAX, thereby promoting apoptosis. Although the regulation of small molecules for PPI interaction is more universal for target positioning, it also has its drawbacks.
Because the interface surface area of PPI interaction is often large (about 1000-3000 Å^2), and the interaction interface of PPI is relatively flat, it is difficult for small molecules and proteins to bind, with low affinity and poor druggability.
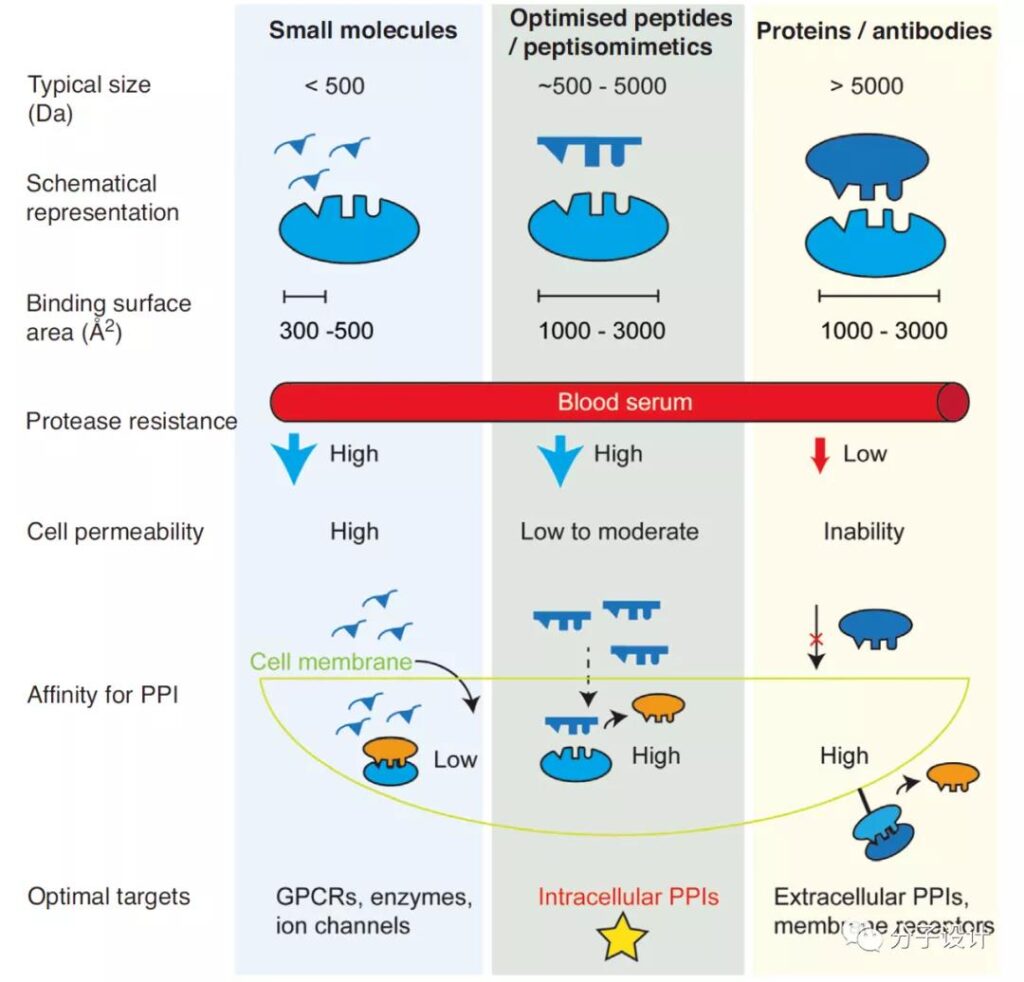
Polypeptides are a class of molecules with high affinity for biology and biophysical properties of small molecules. They are an important strategy for the development and regulation of PPI interaction drugs. However, due to the characteristics of polypeptides, there are still many challenges in converting PPI-targeted polypeptides into therapeutic drugs, such as: serum protease stability and drug resistance; target affinity and toxicity, cell permeability, etc.
In order to improve the druggability of peptides, drug developers often need to use different methods to chemically optimize them (such as cyclization) and limit their secondary structure to a stable and specific conformation to improve biological activity and specificity.
1. Cyclization Strategy (Cyclization Strategy) to stabilize peptides
The cyclization strategy can reduce the binding energy by restricting the conformation of the polypeptide, thereby improving its chemical stability and drug-like properties, and can enhance its affinity for the target.
Chemical cyclization usually stabilizes polypeptides by forming macrocyclic peptides (ie, forming a loop containing multiple amino acid residues).
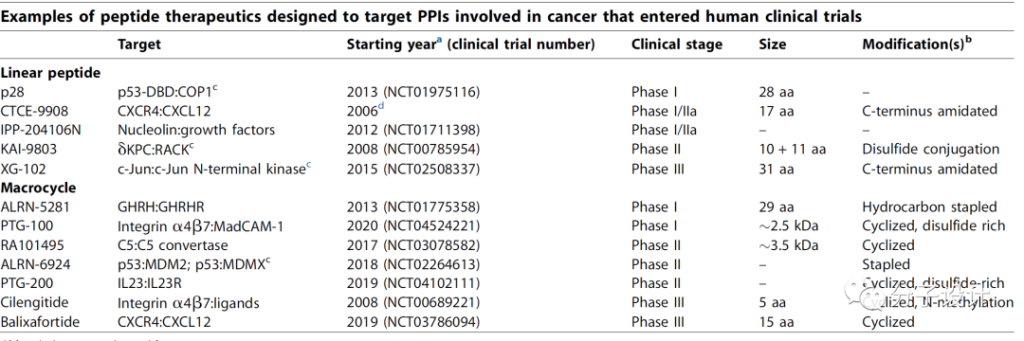
Examples include the immunosuppressant cyclosporine A, the antibiotics vancomycin and polymyxin (usually administered intravenously or inhaled), and the analgesic ziconotide (via intrathecal injection).
A common method in the cyclization strategy is to use “side chain to side chain” cyclization, called Stapling (stapling peptide). In this method, the residues on the adjacent corners of the α-helix are connected by side chain covalent bonds. The classic linkers that can be used in the Stapling method include lactams, alkenes, triazoles, thioethers, and disulfide bonds; after the linear peptide is transformed into a staple peptide, the energy required for its binding to the protein interface decreases and the affinity increases.
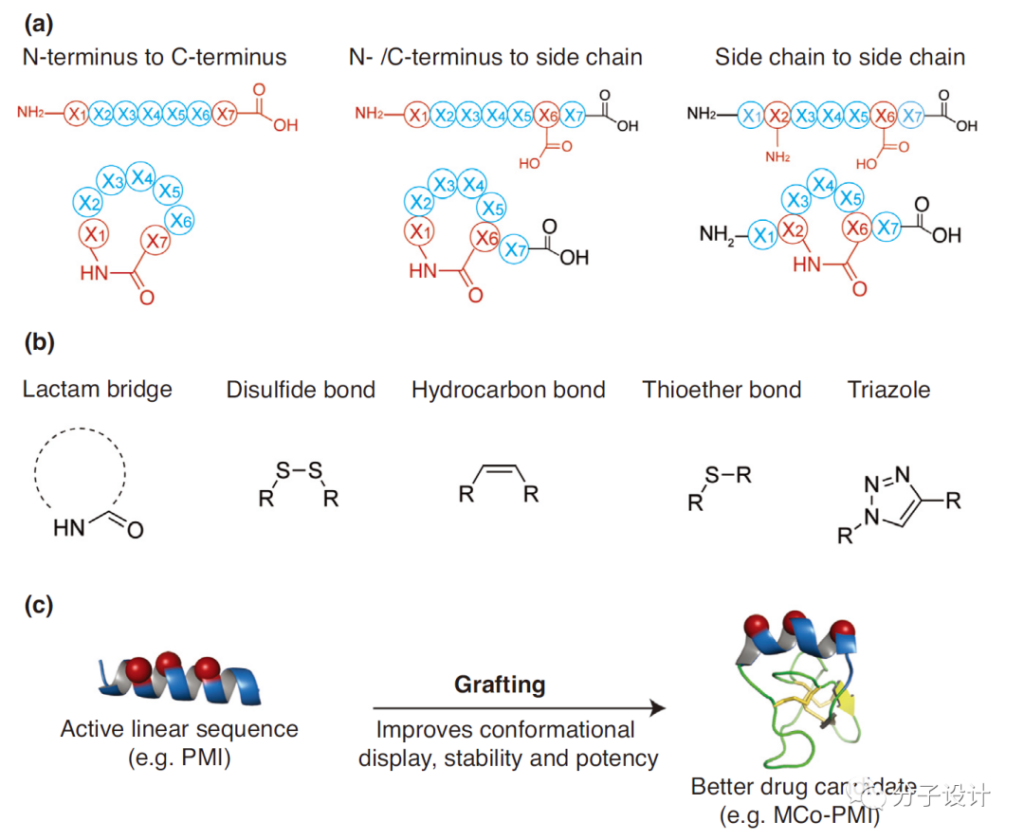
Another strategy of cyclic peptides is to cyclize the peptide bond between the N-terminus and C-terminus of the polypeptide (ie, backbone cyclization), which is called Molecular Grafting. It has been found that a variety of designed macrocyclic peptides can selectively bind to targets involving obesity, cancer and HIV. For example, grafting the active sequence PMI (a mimic of p53) into a cyclic peptide can effectively inhibit the interaction between p53 and MDM2 and MDMX.
2. High-throughput screening to obtain high-affinity peptides
High-throughput screening strategies can be used to identify polypeptides with high binding affinity to the target. The classic screening methods mainly include: Phage display, intein-mediated cyclization of peptides and proteins (SICLOPPS), mRNA display and “One-bead-one-compound” , OBOC)” technology.
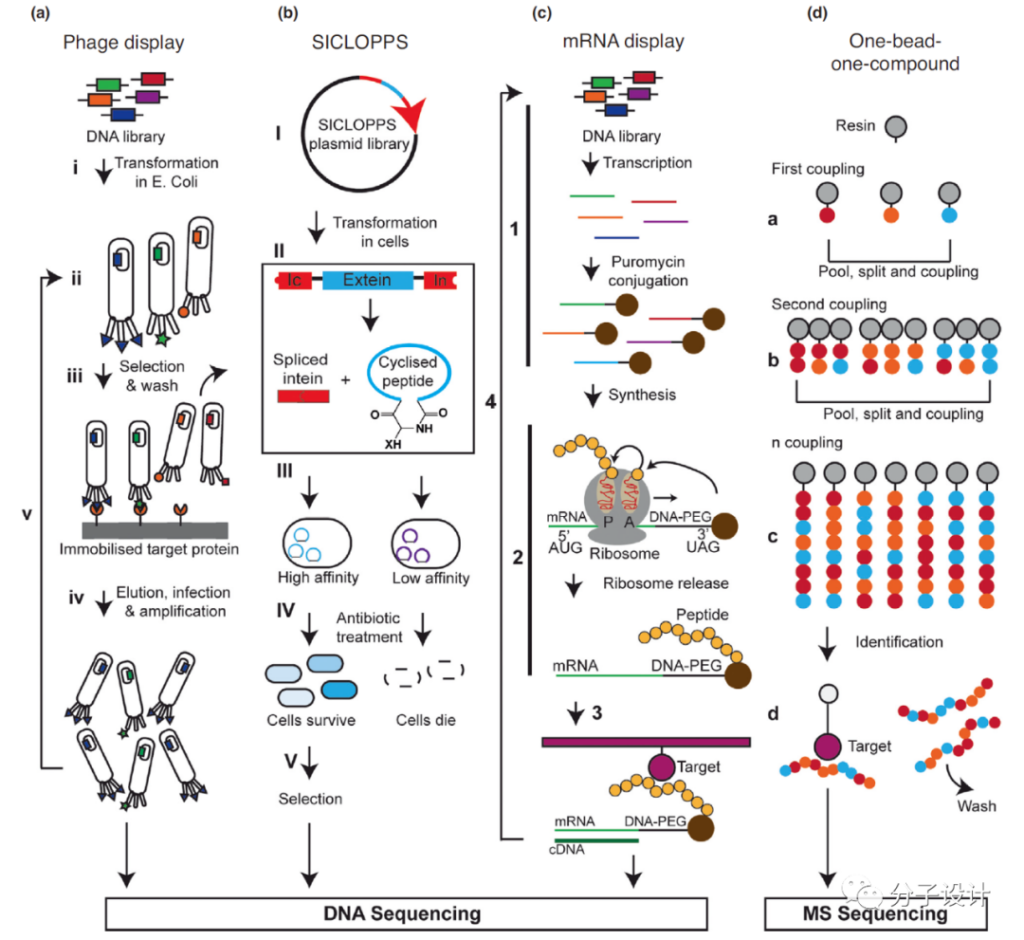
Phage display method is to insert DNA sequence into phage, and phage capsid protein fusion display on the phage surface, high-throughput screening and enrichment to determine the most effective binders; SICLOPPS technology is to use the protein from the protein domain (external protein The ability of) to remove itself and catalyze the connection of the head and tail peptides generates macrocyclic peptides in the cell.
The plasmid encodes a randomized peptide sequence, which is followed by lactam cyclization of the fragment. This technique is usually limited to peptides containing 20 typical amino acids. The mRNA display technology is to form mRNA-protein fusions by covalently binding mRNA and its encoded polypeptide or protein during the screening process to form a large-capacity polypeptide screening library (10^13 ~10^15), and the screening results A polypeptide that can bind to a specific target.
OBOC technology uses magnetic beads to sequentially synthesize D-amino acids (or amino acids with modified side chains) building blocks through chemical methods, and then cyclize them through various chemical strategies and cut them from magnetic beads to establish screening libraries. The target protein on another set of magnetic beads separates effective peptides from the screening library.
3. Penetrating peptide improves the cell permeability of the peptide leader
Cell-Penetrating Peptides (CPPs) have been widely used to carry large proteins and other hydrophilic molecules into cells. There are currently two mechanisms for CPPs to enter cells: ATP-dependent endocytic pathways and ATP-independent membrane translocation pathways.
Most CPPs are positively charged, and their interaction with cell membranes is accomplished by electrostatic interaction with phospholipid head groups and glycosaminoglycans (GAGs) on the cell surface. Currently, the most widely used transmembrane peptide is from HIV-1 transcription transactivator (Trans-activator of transcription, Tat).
Tat is a short positively charged peptide (GRKKRRQRRRPQ) composed of 12 amino acids, which is responsible for carrying and delivering peptide drugs to cells.
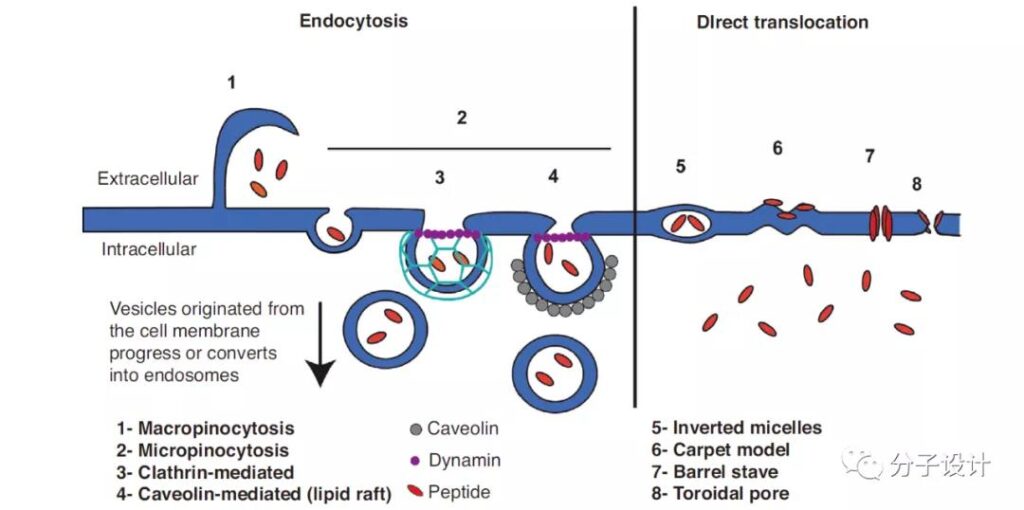
Although thousands of CPPs with penetrating effect have been found, only a few CPP-conjugated drugs have been used clinically.
In order to improve the efficiency of penetrating peptides and the kinetics of their internalization, Hackenberger and Cardoso group cyclized the penetrating peptides, using cyclic peptide R10 (cR10) to convert the nanobody (that is, relative to normal immunoglobulins). , A relatively small single-chain antibody fragment with good stability and solubility) is delivered to the cell cytoplasm.
In addition, by destroying the membrane that wraps CPP endocytic vesicles to release high-content peptides or at the same time improve the ability of direct membrane translocation, both are feasible strategies for optimizing CPP-conjugated drugs into intracellular targets.
Summary:
based on its inherent advantages, peptides are gradually developing into a promising method for the development of PPI target drugs. In order to better develop peptide drugs, the establishment of various biological high-throughput screening technologies and new chemical modification methods have effectively enhanced the resistance of peptides to proteases and facilitated the conversion of peptides into drug lead molecules. In addition, research on the uptake mechanism of CPPs also provides more ideas for the development of peptides targeting PPIs in cells.
(source:internet, reference only)
Disclaimer of medicaltrend.org



