PROTAC: Research trends in the next ten years
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
PROTAC: Research trends in the next ten years
PROTAC: Research trends in the next ten years. Since it was proved in 2001 that the protein degradation targeting chimera (PROTAC) can regulate the function of the protein by holding the ubiquitin protease system (UPS), now PROTAC has gone through the first two decades of rapid development.
The early developed peptide-based PROTAC has poor membrane permeability, and more and more PROTACs are designed based on small molecule drugs for the degradation of membrane proteins and intracellular protein degradation.
The success of PROTAC has promoted the development of the entire targeted protein degradation (TPD) field, and the degradation pathway has also been extended to lysosome LYTACs, autophagy AUTACs and ATTECs. This article focuses on the current research trends of PROTAC, and looks forward to the future.
01 Ubiquitin-proteasome system and “event driven”
In 2004, the Nobel Prize in Chemistry was awarded to Israeli scientists Aaron Ciechanover, Avram Hershko, and American scientist Irwin Rose for their discovery that cells clean up abnormal proteins through Ubiquitin (Ubiquitin, Ub)-regulated protein degradation processes. Ubiquitin is a class of evolutionary conserved small peptides, containing 76 amino acids and a size of about 8.6 kDa. The ubiquitination modification of protein involves a series of reactions of ubiquitin activating enzyme E1, ubiquitin conjugating enzyme E2 and ubiquitin ligase E3. First, under the condition of ATP energy supply, the enzyme E1 adheres to the Cys residue at the tail of the ubiquitin molecule to activate ubiquitin. Then, E1 transfers the activated ubiquitin molecule to the E2 enzyme. The E3 enzyme recognizes the target protein together and modifies it by ubiquitination. According to the relative ratio of E3 to the target protein, it can be divided into monoubiquitination modification and polyubiquitination modification.
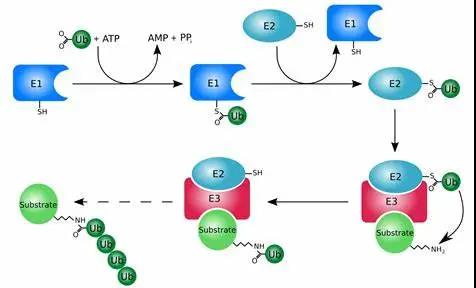
Based on the principle of ubiquitination, medicinal chemists proposed the concept of PROTAC as early as 2001. By designing a ligand that binds to the target protein at one end, and a ligand that binds to the E3 ubiquitin ligase at the other end, connect with a linker to draw the target protein and E3 enzyme closer in the body, so that the target protein is labeled with ubiquitin, and then Degraded through the ubiquitin-proteasome pathway. Unlike the “occupy drive” of small molecule inhibitors and antibodies that need to occupy the active site of the target protein to block its function, the degradation of the target protein by PROTAC is “event driven”, that is, it is only responsible for triggering the E3 enzyme and the target protein. Binding, no space-occupying, and no need to inhibit the target protein. Therefore, PROTAC has great potential for targets without clear active pockets and intracellular targets that cannot be accessed by macromolecular antibodies.
The ability of PROTAC to degrade the target protein is related to the ternary complex formed transiently by the “target protein-PROTAC-E3 ligase”. Studies have found that although sometimes the ligands used for targeting proteins have low affinity, as long as they can form a ternary complex, the designed PROTAC still has a good degradability. For example, the Crews laboratory once designed PROTAC for p38α kinase. Although Foretinib has an affinity for p38α kinase at the level of 10 μM, the designed PROTAC has a better degradation effect on p38α kinase (DC50=210 nM). After PROTAC induces the ubiquitination of the target protein, it quickly dissociates and participates in the formation of the next ternary complex.
02 Degradation of difficult target proteins
PROTACs have made phased progress on targets that were difficult to be medicines in the past, such as scaffold protein FRS2α (2013), protein complex BAF (2019), transcription factor STAT3 (2019), Ras family protein (2020), etc., and will also make progress in the future. The degradation of more non-drugable targets becomes possible.
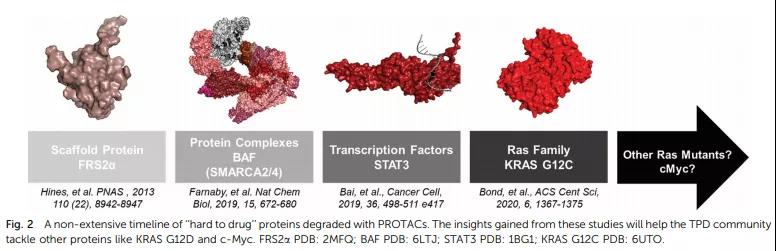
FRS2α is an important scaffold protein in the process of neural differentiation and participates in the MAPK pathway mediated by nerve growth factor NGF. The Crews project tried to design a membrane-permeable peptide chimera TrkAPPFRS2a as early as 2013. The structure contains the sequence recognized by FRS2α and the sequence recognized by the E3 ligase VHL, but its concentration is as high as 50 μM or more, and the effect is not exhausted Pleasant.
Chromatin is a carrier of eukaryotic genetic information, and the stable regulation of its structure is essential for many biological processes such as DNA replication, transcription, and gene damage repair. The ATP-dependent chromatin remodeling complex BAF/PBAF changes the position and composition of nucleosomes through slipping and elimination to achieve dynamic regulation of chromatin structure. About 20% of tumors are mutated with this complex. closely related. The study found that two ATPases in the complex: SMARCA2/4 are potential cancer treatment targets. The researchers connected the SMARCA2/4 bromodomain inhibitor to the VHL E3 ubiquitin ligase ligand through a linker to form a PROTAC molecule that successfully degraded SMARCA2/4, and proved that it has an anti-tumor proliferation effect in AML cell lines. This result verifies the possibility of targeted degradation of the complex.
The development of drugs that target transcription factors is often troubled by membrane permeability and the large protein-protein interaction (PPI) surface area of the complex, which makes it difficult to prepare drugs. As early as more than ten years ago, small molecules that can target the SH2 domain of STAT3 have been reported, but due to their low efficacy and lack of selectivity as inhibitors, they did not solve practical problems. Recently, the PROTAC molecule SD-36 formed by linking STAT3 targeting molecule SI-109 and lenalidomide can successfully degrade STAT3 protein in leukemia cell lines and induce cell death. What’s more interesting is that the selectivity of SI-109 molecule, which was originally not outstanding, is also improved after being designed as a PROTAC degradant.
Mutations in the RAS protein occur in 25% of tumors and are a promising target. However, due to its lack of effective pockets and high affinity for GTP, it has been used as a “non-drugable target” for a long time. In recent years, covalent inhibitors designed for KRAS G12C mutations provide a new idea for targeting RAS mutations. Representative drugs AMG 510 and MRTX849 have all entered the clinical stage. However, the use of KRAS G12C inhibitors often causes tumor cells to develop tolerance through other ways. Therefore, it is necessary to develop targeted degradation of RAS protein. The Crews research group tried to use MRTX849 as a small molecule targeting KRAS, synthesized a series of PROTACs, and identified effective LC-2. LC-2 can rapidly and continuously degrade KRAS G12C with a DC50 of 0.25–0.76 μM. Further research found that LC-2 could not inhibit the pathway downstream of KRAS, and compared with MRTX849, the anti-tumor proliferation effect was not enhanced. The reason may be that the use of MRTX849 covalent inhibitor makes the catalytic properties of PROTACs lost.
03 Looking for small molecule ligands for designing PROTACs
Finding novel ligands for target proteins is a major core of PROTAC technology. Chemical-proteomics competition experiment and DNA-encoded compound library (DEL) technology are respectively advantageous screening methods for discovering covalent and non-covalent binding ligands. Among them, the DEL technology is currently being done by the domestic Chengdu Leading Pharmaceutical Industry. The relevant technical principles can be found in an article on other public accounts, and I will not repeat it here (click on the link).
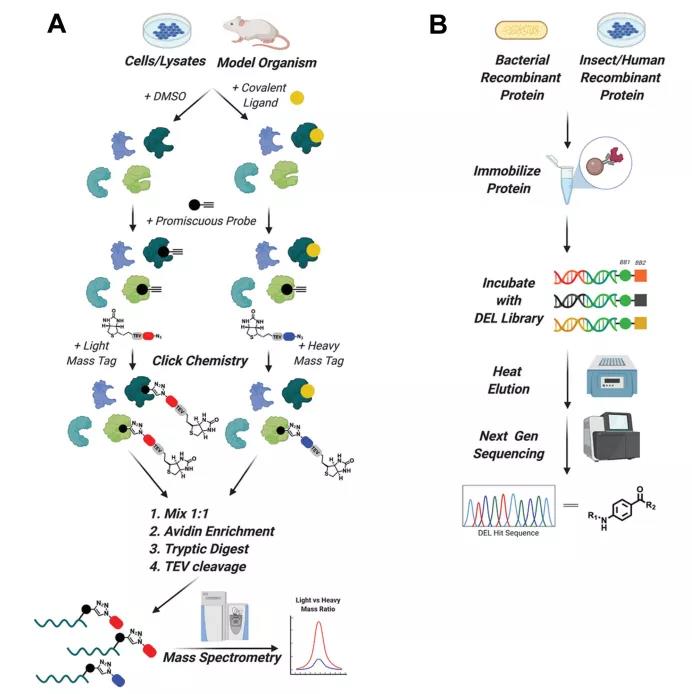
The basic flow of the chemical-proteomics competition experiment is shown in the figure: Whole cells containing protein, cell lysates and even animal tissues are incubated with the compound to be tested, and the DMSO group is used as a control. After the end, it is then combined with a pan-specific covalent binding agent with an alkynyl tag (for example: iodoacetamide that can recognize Cys). Then, label the Click with a large molecular weight in the sample group, and label with a small molecular weight in the Click group in the control group. After mixing the two groups of samples 1:1, avidin enrichment, trypsin digestion, TEV fragmentation of Biotin-AV. Purified proteins with tags of different molecular weights are identified by mass spectrometry. A larger light/weight ratio indicates that the protein is covalently bound to the compound, and the protein is a potential target for covalent binding. A light-to-weight ratio of 1 means that no compound is covalently bound. Using this scheme, Nomura’s research team successfully identified the covalent ligand EN4 for Myc target.
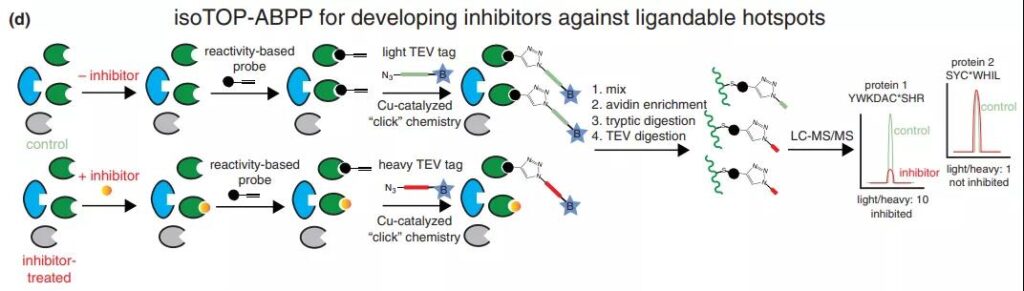
04 Develop more E3 ubiquitin ligases that can be used for recruitment
Compared with finding small molecule ligands for POI, finding the ligands of E3 ubiquitin ligase is actually more difficult and more accidental. There are two main reasons: one is that there is not much knowledge about the structure of E3 ligase; the other is The small molecule we found must be able to target E3 ubiquitin ligase without affecting its activity. Therefore, the E3 ubiquitin ligase currently used mainly still uses the classics (such as VHL, CRBN, MDM2, clAP, etc.), but with the deepening of the understanding of the relative role of E3 ubiquitin ligase substrates and the advancement of screening techniques, I believe more Ligands that can recruit E3 ubiquitin ligase will be discovered.
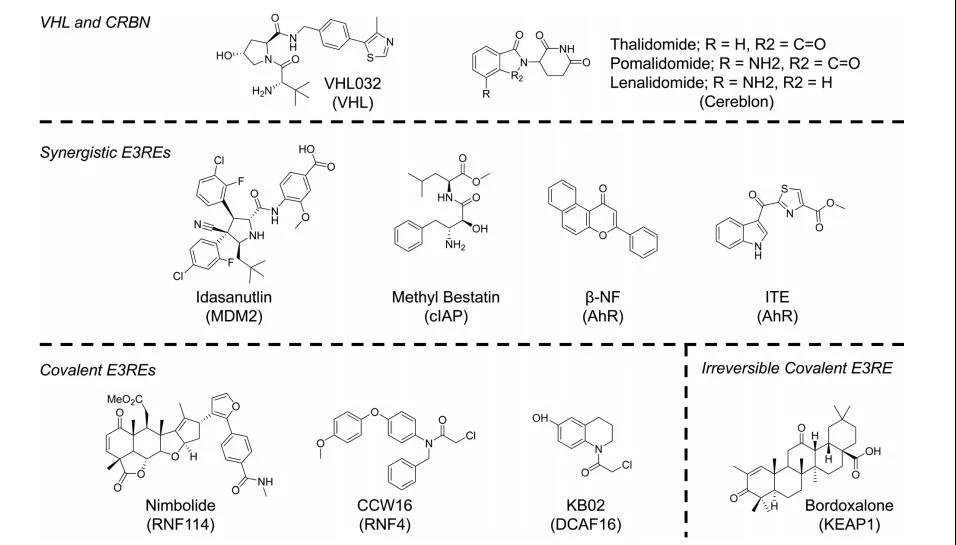
The figure above is the current ligand structure used to recruit different E3 ubiquitin ligases. CRBN and VHL are the most commonly used E3 ubiquitin ligases. Among them, thalidomide, pomalidomide, and lenalidomide have been widely used to design and recruit the targeted degradation of CRBN ubiquitin ligase. In the early stage, the ligand used to recruit VHL was a peptide that mimics its natural substrate HIFα. Later, researchers discovered that the weak affinity ligand (10 μM level) of VHL can also be effectively used for targeted degradation. It has been reported that degradation The activity can even reach the nanomolar level. This phenomenon may imply that the high affinity of E3 ligase ligand and E3 ligase is not necessarily necessary.
Ubiquitin ligases MDM2, cIAP, and AhR have been used in the field of targeted degradation for many years, but compared with CRBN or VHL, these ubiquitin ligases are currently not widely used. In recent years, with the discovery of more and more MDM2-p53 protein-protein interaction inhibitors, the targeted degradation of the remaining MDM2 ubiquitin ligase has also been further developed. It is worth noting that PROTACs designed based on cIAP, MDM2, and AhR can not only degrade oncoproteins, but also induce apoptosis by degrading cIAP, up-regulating p53, etc., and synergistically increase the effect of anti-tumor cell proliferation.
It has also been reported to recruit DCAF16, RNF114, and RNF4 with ubiquitin ligase function through covalent small molecule ligands for targeted protein degradation. Nimbolide is a naturally extracted triterpenoid compound with pharmacological effects in preventing and treating tumors. Exploring its mechanism, it is found that it can be combined with C8 of RNF114. The PROTACs designed by researchers with nimbolide and BRD target inhibitor JQ1 have nanomolar degradation activity on BRD4, and can exert a synergistic effect similar to MDM2 by stabilizing tumor suppressor factors such as p21. Although good experimental results have been obtained, but limited to the natural source of nimbolide, finding a synthetic alternative ligand for RNF114 is the key to its wide use. PROTACs developed with DCAF16, RNF114, and RNF4 not only expand the available E3 ubiquitin ligase types, but also prove that PROTACs that covalently bind E3 ubiquitin ligase can also have catalytic properties.
PROTACs designed based on POI reversible covalent ligands have been successfully verified on BTK targets. In order to further verify whether it is possible to design PROTACs based on the reversible covalent ligand of E3 ubiquitin ligase, the researchers linked bardoxolone with reversible α-cyanoenone hetero-Michael acceptor to JQ-1 to design PROTAC The molecule CDDO-JQ1 successfully degraded BRD4 by recruiting KEAP1, which also verified its feasibility.
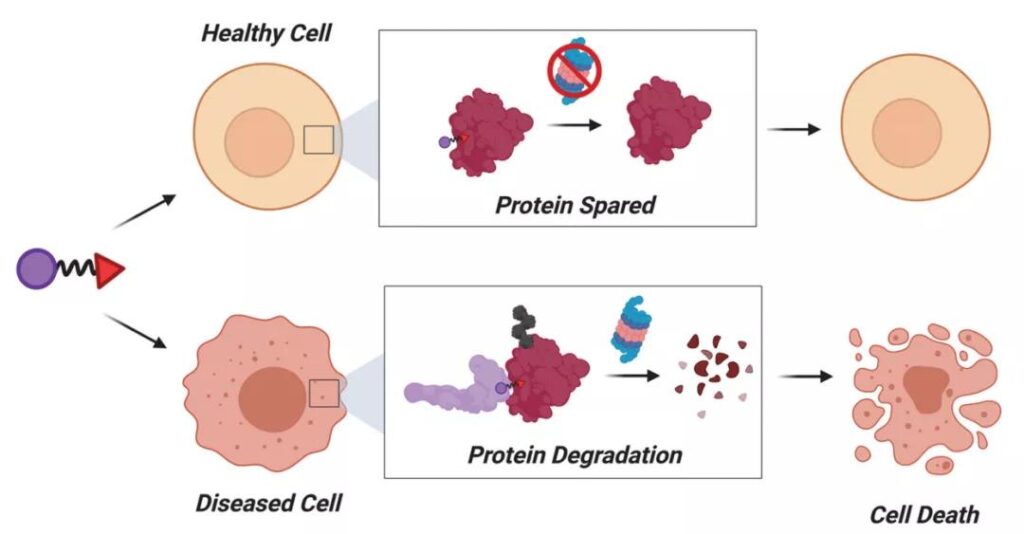
The indistinguishability of PROTAC between the diseased site and the normal site of POI is a major hidden danger that leads to its systemic toxicity. Studies have shown that there are no less than 600 E3 ubiquitin ligases in the human proteome, some of which are tissue and disease specific. In the next stage of PROTAC development, if the distribution characteristics of E3 ubiquitin ligase can be used to improve the tissue/disease selectivity of PROTAC degradation, the safety of PROTAC will be improved.
A classic example is: BCL-XL is an anti-apoptotic protein related to BCL2, which is essential for platelet survival. While inhibiting BCL-XL, navitoclax, the BCL-XL inhibitor, can cause dose-limiting thrombocytopenia by degrading platelet BCL-XL, which limits its clinical use. Considering the low expression of E3 ubiquitin ligases such as VHL, CRBN, and cIAP in platelets, researchers at the University of Florida and MD Anderson Cancer Center used these E3 ligases to design targeted degradation of BCL-XL PROTAC selectively degrades BCL-XL in tumor cells and reduces platelet toxicity. For more information about the tissue specificity of E3 ubiquitin ligase, please refer to the HμMan Protein Atlas database (https://www.proteinatlas.org/).
05 Improve the efficiency of PROTAC discovery
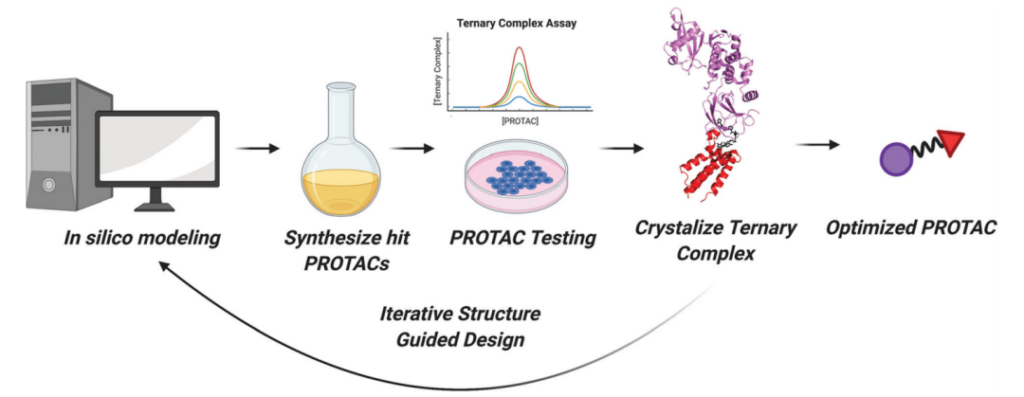
In the past 20 years, although PROTAC has made great progress, the main discovery methods have not been significantly innovative: the preliminary design of compounds by changing the linker type, length, ligand type, and connection position, through cell experiments and Wsetern experiments To determine the activity of protein degradation, obtain an active lead compound, and then cycle multiple times to optimize the activity. This iterative optimization method is time-consuming and labor-intensive. If the structure-based design ideas used in the design of small molecule inhibitors can be used for reference in the next development process of PROTAC, the discovery process of PROTAC will be accelerated.
5.1 PROTAC design based on ternary composite structure
Previous studies have shown that the formation of ternary complexes is extremely important for the degradation of target proteins. As long as PROTAC forms a strong ternary complex with POI and recruits E3 ubiquitin ligase, even PROTAC weakly binds to POI (>1 μM). POI degrades. In 2017, researchers reported the first case of the ternary complex structure formed by PROTAC molecule MZ1 with VHL and BRD4BD2. The results showed that MZ1 caused VHL and BRD4BD2 to form a new protein-protein interaction (PPI). ITC experimental results show that the formation of ternary complexes mediated by MZ1 has a positive synergistic effect, and the positive synergistic effect is related to the production of PPI. In addition, the existence of linker can promote the formation of PPI by bringing VHL and BRD4BD2 closer.
In the past few years, the structures of more than ten ternary complexes have been resolved. The common feature is the emergence of new PPIs and the existence of flexible chains that make POI and E3 ligase close together. Further studies have found that reducing the flexibility of the linker by regulating the PPI of the ternary complex may affect the selectivity and effectiveness of PROTAC for POI. With the discovery of more rigid PROTACs, biophysical methods can be used to compare the binding kinetics of flexible and rigid PROTACs to understand how rigidity affects the association/dissociation of the established E3 ligase/POI ternary complex.
5.2 Molecular modeling and calculation-based PROTAC design
The research on PROTAC ternary complexes not only provides ideas for structure-based drug design, and provides a basis for revealing the molecular mechanism of ternary complex formation, but also makes it possible to design computer programs to predict the formation of PROTAC ternary complexes. At present, mathematical models used to characterize the formation of ternary complexes have been reported, which can predict the maximum concentration, association and dissociation kinetics of the ternary complexes formed based on affinity in an equilibrium state.
However, the actual degradation process is not statically balanced, but a continuous flow process. Therefore, in order to solve the problem that the above model cannot accurately describe the degradation kinetics of rapid protein turnover and deubiquitination, the researchers developed a dynamic correction model of TPD molecules based on the Sim-Biology module of MATLAB. This model can predict the effect of PROTAC’s membrane permeability on DC50 and DMax. It can also predict the selectivity based on the affinity of PROTAC to E3 ligase and POI and the stability of the ternary complex. It can even predict the PK and PD of PROTACs. Features are predicted. In addition, program development for PROTAC molecular docking and modeling of ternary complexes has also been carried out based on the MOE or Rosetta platform.
06 Discussion and outlook
It has been twenty years since PROTAC has been developed. With the development of proteomics, sequencing technology, combinatorial chemistry and other technologies, the discovery standard process of PROTAC will also have profound changes. At the end of the article, the author of this article first looks forward to the future discovery process of PROTAC. There are two points worth noting: 1. Computer simulations are used to predict the generation of PPI and select suitable Linker scenarios, such as: AlphaFold, ProsettaC, dynamic simulation And so on will be used in the preliminary design of PROTAC. 2. Advances in structural biology technology (X-Ray, cryo-electron microscopy, etc.) will provide more understanding of the structure of the three original complexes.
In addition, the author of this article believes that the integration of PROTAC and other fields of technology (such as immunotherapy) will change the landscape of the TPD field. Studies have shown that the peptides produced by PROTAC degrading POI can be presented as antigens to MHC receptors, enhancing the effect of immunotherapy. In addition, the combination of PROTAC and immune checkpoint inhibitors also showed a synergistic effect.
In terms of delivery methods, PROTACs can learn from ADC drugs. In addition, nanotechnology and prodrug technology will also contribute to the targeted delivery and metabolic stability of PROTAC.
In addition to the above, in the next stage of the development of the TPD field, the following directions are also worthy of attention: 1. RNA-PROTACs designed based on small molecule RNA mimics are used to degrade RNA binding proteins; 2. Targeting by ribonuclease Chimeras (RIBOTACs) degrade RNA; 3. TRAFTAC technology that uses DNA as a recruiting element to degrade transcription factors; in addition, covalent molecular glues based on natural products and small molecules that can induce BCL6 to polymerize and degrade should also receive sufficient attention.
(source:internet, reference only)
Disclaimer of medicaltrend.org



