Process and quality control of vector for gene modification
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Process and quality control of vector for gene modification
Process and quality control of vector for gene modification. CAR-T cell products are the product of the combination of gene therapy and cell therapy technology.
As a tool for gene therapy, vectors may bring risks such as insertion mutations, replicating viruses, and contamination by foreign factors. At the same time, the genetic material they carry is an important part of CAR-T cells, enabling T cells to have powerful recognition and killing.
An important basis for tumor cell activity. Therefore, the quality of the carrier is very important. It is completely different from other raw materials used in production and should be managed as an integral part of the product.
The vector for gene modification should be produced under cGMP conditions, with high-quality clinical-grade products that have undergone comprehensive quality testing.
The production of viral vectors should establish a control system for the entire process, including raw materials, production processes, quality control of key points, and quality control of viral vectors, etc., and verify the robustness of the process and batch-to-batch consistency.
In addition to meeting the general technical requirements for the production of biological products, the production of viral vectors has certain particularities in terms of raw materials and production process strategies, which should be paid attention to in the process of process development.
1. Key raw materials
The key raw materials usually required for vector production are cells, culture medium and serum, and plasmids. Each of these must come from an approved supplier, and should undergo rigorous testing procedures to reduce the risk of introducing foreign factors into the production process.
At present, the production of lentiviral vectors mainly adopts the third-generation plasmid packaging system. Although the second-generation packaging system has not been reported to produce replicable viruses, the third-generation packaging system is safer in terms of recombination probability and component design.
The construction of γ-retrovirus stable toxin-producing cell lines often adopts a three-plasmid system. The packaging plasmid is used as the raw material for the production of virus vectors, and its quality and batch-to-batch consistency have a direct impact on the yield and quality of the virus. Plasmid production should comply with GMP standards, establish a secondary or tertiary engineering bacterial bank in accordance with the requirements of the Chinese Pharmacopoeia, and complete the corresponding bacterial bank verification.
In addition to meeting the requirements of general recombinant biological products for plasmids, according to the purpose of packaging plasmids, attention should also be paid to the recombination safety of the plasmid element design of the packaging system, the effect of plasmid homogeneity on the efficiency of virus packaging, and the level of residual impurities on the virus and CAR -The impact of T cell quality and the residual level of lactam antibiotics used in production poses a safety risk to allergic individuals.
The cell line often used for lentivirus packaging, such as HEK293T, etc., can be prepared and verified by referring to the requirements of the Chinese Pharmacopoeia for the production of recombinant biotechnology products or cell substrates for vaccines. In addition to the general inspection items, the verification of the cell bank should also include the detection of species-specific internal and exogenous factors to evaluate the safety of recombination during the virus production process.
The production of γ-retrovirus involves transient viral packaging cells (such as HEK 293T) and the final screened stable toxin-producing cell strain (such as PG13). Due to differences in production systems, in addition to the requirements for packaging cells and toxin-producing cell substrates, the production of γ-retrovirus should also pay attention to the control during the construction of stable production cell lines and the final verification of stable toxin-producing strains, such as biological sources. The application of raw materials, the identification of gene sequences, the risk of exogenous factors, the screening of cell monoclonals, the risk of recombination in the process, etc. In addition, attention should also be paid to the risk of producing GaLV-like viruses during PG13 cell culture and production.
2. Virus production process
Based on the concept of QbD (quality by design) and the awareness of risk control, the virus production process should be rationally designed. Through the entire process of quality control, the consistency of the virus batch should be ensured, the residual impurities, and the variability of CAR-T cell production should be reduced.
The production process of the carrier must be verified to prove that it can repeatedly produce a safe, effective and consistent quality carrier with a controlled process flow.
γ-retrovirus is usually obtained from the recovery and culture of stable toxin-producing cell lines. The stable transgenic cells for packaging retrovirus need to establish a cell bank system. For the production of γ-retrovirus, attention should be paid to the process of constructing toxin-producing cell lines to produce replication The risk of virus.
The method of adherent culture of HEK 293T, PG13 and other cells also poses certain challenges to scale-up. Although at present, spinner flasks, multi-layer shake flasks, cell factories, hollow fiber reactors, fixed-bed bioreactors, and microbead carriers have been developed. And many other culture strategies to increase the surface area, but the cell density and space utilization of these culture methods are still low.
Because the envelope protein of γ-retrovirus is more sensitive to shearing force and other effects, it is usually not suitable for conventional purification methods, and membrane filtration is generally used for purification. At the same time, the biological raw materials such as fetal bovine serum that are often added during the culture process are easy to introduce exogenous factors, which further increases the challenge of the purification process.
The production of lentivirus usually adopts the transient packaging method, because of the cytotoxicity of viral proteins such as protease, gag-pol, VSV-G, and the transiently transfected lentiviral vector preparation system requires the establishment of a strain library and a cell library system. It needs to be managed and tested in accordance with the corresponding requirements.
Established cell banks, virus seed banks, bacterial seed banks, and animal-derived media components also require extensive testing. Moreover, the large batch-to-batch variability of this production method and the large demand for plasmids limit the scale of production to a certain extent.
The development of a medium with limited chemical composition for suspension culture is still a relatively ideal culture method, which is not only conducive to scale expansion, but also avoids the use of high-risk biological raw materials such as serum, and reduces the burden of downstream purification.
The virus packaging process should optimize and verify the cell culture conditions, culture time, transfection method, plasmid dosage, harvest time and frequency and other processes through process research to ensure the consistency of virus yield and quality between batches.
At present, the commonly used purification methods for the production of lentiviral vectors include ultracentrifugation, ion exchange chromatography, molecular exclusion chromatography, affinity chromatography, and diafiltration. In order to obtain higher virus purity and concentration, it is necessary to rationally design the purification route and optimize the combination of one or more purification units according to the structural characteristics of the virus, the type of host cell, the composition of the medium, the nature of the purification method, etc. Improve the purity of virus products and reduce the safety risk of impurity residues.
Due to different purification strategies, the types and purity of impurities in virus products are also different. While removing process and product-related impurities, virus purification should maintain the structural integrity, infectious activity, and expression function of the target gene as much as possible.
Since the cell lines used in the construction of toxin-producing cell lines, such as PG13, HEK 293, etc., have a certain degree of tumorigenicity, the remaining tumor-forming risk cells, cell debris and other impurities should be removed through the purification process, and at the same time should be reduced or The serum-free culture process reduces the residual amount of biological raw materials such as fetal bovine serum in virus products [21].
3. Quality research and control
For retroviral vectors, lentiviral vectors] and transposon plasmid systems, the usual test items for quality control of the final vector product are shown in the table below. The quality control items and standards of different carriers should be established according to their own characteristics and production process conditions.
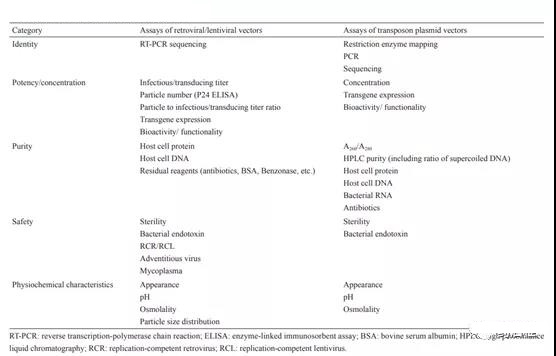
In addition, because the genetic modification vector of CAR-T cells does not directly enter the patient’s body, but undergoes steps such as dilution and washing along with the cell process, it is necessary to formulate reasonable quality standards in accordance with process verification and risk assessment. Virus products The residual limits of various impurities should ensure that they can be stably removed or reduced to a safe human dose level through the liquid replacement and washing process planned for CAR-T cell production. In addition, in order to reduce the risk of genotoxicity and tumorigenesis, you can refer to the FDA’s recommendation that the size of residual DNA fragments should be less than 200bp.
Central control points: The production of replicable viruses is the most serious risk factor in the production and application of lentivirus and γ-retroviral vectors. The cells and supernatants of the master cell bank should be used to produce terminal cells and harvest fluid. Supernatants, final virus products, and later cell products are monitored and controlled to ensure that there are no replicable viruses.
At the virus stage, the detection of replicable viruses should adopt a cell-based method, and the setting of the RCR/RCL standard limit should take into account the amount of virus in the subsequent process and the detection limit of the methodology.
In addition to improving the sensitivity and detection limit of the methodology, a variety of complementary detection methods can be considered, such as P24 immunological methods and PCR methods, VSV-G immunological methods and PCR methods, and reverse transcriptase activity Quantitative analysis, etc.
In addition, appropriate representative controls or reference standards should be selected to verify the methodology. For details, please refer to the latest RCR testing methods and gene therapy guidelines of the US FDA.
In addition to evaluating the promoter/enhancer elements of the viral genome, potential splicing sites, etc. during the virus design stage, the differences in host cells can be determined by high-throughput sequencing and other methods for a certain number of target transfected cells. The tendency of the virus insertion site is monitored, and the safety of the virus insertion site is preliminarily assessed. At the same time, the representative patients using CAR-T cell products should be dynamically tracked and monitored and regular and long-term follow-ups should be carried out to rule out the virus insertion. For adverse reactions, please refer to the FDA guidelines for patient follow-up.
4. Stability and compatibility
The carrier harvested during the production process should be purified and stored under an appropriate formulation. At the same time, stability studies should be carried out to ensure the stability of the carrier product.
(1) Compatibility
The production process of viral vectors involves the contact between production pipelines and packaging containers, and attention should be paid to the quality standards and compatibility of the directly contacted materials, and the impact of material adsorption and exudation should be evaluated.
At the same time, since the storage stability of virus products is directly related to transfection efficiency and degradation risk, the factors affecting virus storage should be comprehensively evaluated, especially the effect of freezing and thawing conditions on virus titer. Specifically include the following:
(2) Long-term stability, storage conditions such as -80℃, set reasonable time points and test items;
(3) Freeze-thaw stability
(4) Stability during use, such as 4°C, room temperature, etc.;
You can refer to the relevant guidelines for the stability of biological products, combined with the product quality research results and release testing items, select sensitivity indicators for observation (usually titer, purity, etc.), summarize important influencing factors and quality change trends, according to virus stability The results of the study reasonably determine the validity period of virus preservation.
(source:internet, reference only)
Disclaimer of medicaltrend.org



