What is ELISA and its Development Optimization and Improvement?
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
What is ELISA? Development, Optimization and Improvement?
What is ELISA and its Development Optimization Improvement ? Enzyme-linked immunosorbent assay (ELISA) is an immunological technique used to detect and measure specific proteins, such as antibodies, antigens, and hormones in biological samples.
How does ELISA work?
ELISA is based on specific antigen-antibody reactions and usually involves immobilizing antibodies or antigens to 96-well or 384-well plates. The basic steps of ELISA:
- Fix the target protein/antigen on the surface of the microplate
- Wash unbound/excess protein/antigen from the plate
- Add labeled antibody, which will then bind to the target antigen/protein present in the plate
- Wash out unbound (excess) antibody from the plate
- Add an enzyme-specific substrate, which will react with the enzyme and produce a colored product, which can be colorimetrically determined using a microplate reader.
Horseradish peroxidase (HRP) or alkaline phosphatase are commonly used enzymes in ELISA, and the substrates include tetramethylbenzidine (TMB) and 2,2′-azido-bis-3-ethylbenzene And thiazoline-6-sulfonic acid (ABTS). Usually select repeated or three sampling, and use different concentrations of samples to ensure a biologically acceptable detection range. As shown in Figure 1.
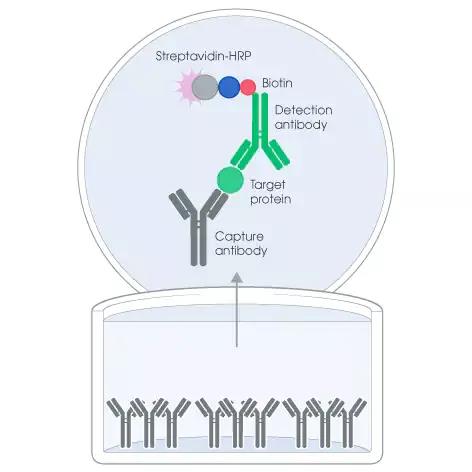
Figure 1. Basic schematic of ELISA analysis.
Standard curve line
In order to quantify the concentration of the target antigen, a standard curve is generated using an antigen of known concentration. Next, the optical density (the light absorption of the enzyme-substrate reaction product) obtained from the colorimetric determination is plotted on a standard curve to accurately measure the level of the target antigen in the biological sample.
Types of ELISA
Direct ELISA
Fix the target protein/antigen on the surface of the plate, and then add the enzyme-labeled antibody produced against the target molecule. After adding a suitable substrate, perform colorimetric detection. This format is simple and time-saving. However, because all target antigens are bound to the surface and the difficulty of primary antibody labeling, there is a high experimental background.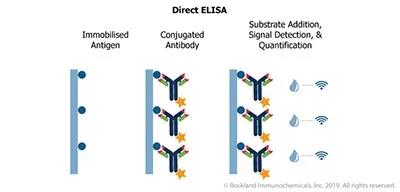
Indirect ELISA
The target protein/antigen immobilized on the surface of the plate is incubated with the primary antibody raised against the target molecule. Then, the enzyme-labeled secondary antibody generated against the primary antibody is used for detection and quantification. Although this format is more sensitive than direct ELISA, it has a high false positive detection due to the crossover of the secondary antibody.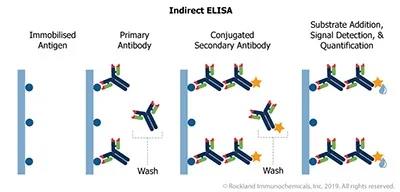
Sandwich ELISA
This ELISA requires two antibodies (capture antibody and detection antibody) generated against different epitopes (specific antibody binding sites of the antigen) of the target protein/antigen.
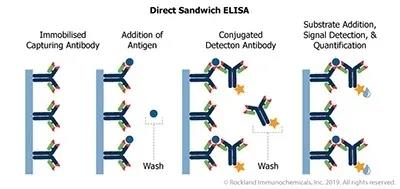
The procedure includes immobilizing the antibody against the target antigen (capture antibody) in the microplate; adding the biological sample containing the target antigen to the plate, and then combining with the immobilized antibody present in the plate; adding the enzyme-labeled antibody (detection antibody) The antibody will then bind to the target antigen present in the plate; and the enzyme-specific substrate will be added to the plate, which will react with the enzyme and produce a colored product for detection. This format involves two antibodies that detect different epitopes of the target molecule, making it very specific.
Competitive ELISA
In this process, the reference antigen is immobilized on the surface of the plate, and a biological sample pre-incubated with a specific amount of labeled antibody is added to the plate. The amount of antigen present in the sample will determine the amount of unbound or free antibody that can be used to bind the reference antigen in the plate. This format is particularly suitable for low molecular weight targets.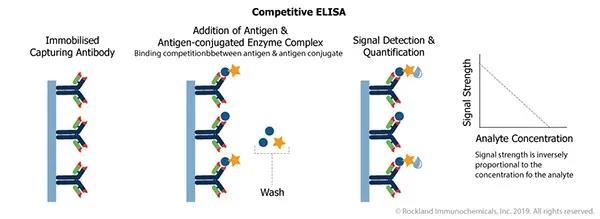
What are the advantages of ELISA?
The main advantage of ELISA is its high sensitivity and specificity, and it is suitable for detecting target molecules even at pictographic level. Because the experimental procedure is simple and not demanding, it is often used for high-throughput screening.
ELISA can also be used to quantify target molecules in various samples, including serum, plasma, urine, saliva, cell or tissue extracts, etc.
What are the disadvantages of ELISA?
Since quantification is based on an enzyme-substrate reaction, the detection time frame is very short. In addition, only a limited amount of information can be obtained by ELISA, such as the presence or number of target molecules. This technique cannot obtain information related to molecular activity.
ELISA applications
Cancer screening, medication and pregnancy testing
The identification of cancer biomarkers for early detection of cancer is a challenge that is constantly being developed and researched. ELISA-based technology is available and clinically used to detect early cancers-including ovarian cancer and breast cancer.
In addition, ELISA can be used to determine the concentration of illegal drugs in urine samples, such as cannabinoids, amphetamines, opioids, cocaine, benzodiazepines, and methadones. This method can also be used to monitor drug concentration levels in patients receiving treatment, such as anti-drug antibodies in patients with rheumatoid arthritis and inflammatory bowel disease.
ELISA is also often used to detect human chorionic gonadotropin (hCG) in urine, which is higher in pregnant women, so it can be easily accessed and tested at home.
Detection of platelet antibodies
The detection of platelet antibodies in serum is used to identify patients with diseases such as idiopathic thrombocytopenic purpura and systemic lupus erythematosus. These tests use alkaline phosphatase, which is labeled anti-human IgG.
In addition to providing a cheaper and complex method for detecting platelet antibodies, ELISA has also been found to provide more functions than other commonly used detection methods such as lymphocyte toxicity (LCT) and platelet immunofluorescence detection (PIIFT).
Food allergens
ELISA is widely used in the food industry to detect the presence of allergens and to label ingredients required by law. This application greatly benefits from the sensitivity of ELISA and can detect levels of potential food allergen contaminants at low parts per million (ppm) levels. It also has the advantage of being able to test for oil and other substances, such as egg white or milk, which cannot be detected by alternative methods such as PCR.
Use viruses to detect viruses
The advantage of using ELISA to detect viruses is that they can be used in developing countries where infection rates are usually high, and can reach the most vulnerable population through on-site testing capabilities and instant results, allowing opportunistic testing (for example, in emergency departments).
HIV:
ELISA is the first universal HIV test kit that detects positive patients by testing human serum Cystatin C.
West Nile virus:
West Nile virus is tested by IgM antibody capture (MAC) ELISA in the patient’s serum or cerebrospinal fluid (CSF). The test is performed 8 to 21 days after the onset of symptoms. The test can also confirm whether the infection has progressed to the patient’s central nervous system.
Newcastle disease virus (NDV):
NVD is an avian virus that can be transmitted to humans. Depending on the strains present, the severity of NDV disease may vary, from moderate respiratory dysfunction to diarrhea and other life-threatening symptoms. From the deadliest NDV strains (rapid-onset NDV) to increasingly less severe strains (mid-source NDV and chronic source NDV), ELISA is used to monitor their presence in the population, help coordinate vaccination plans, and identify Any flock of chickens infected with NDV.
ELISA development skills
Because of its flexibility, sensitivity and scalability, ELISA is one of the most widely used immunoassay techniques. During the ELISA development process, several key factors must be optimized to ensure accurate results. Here, we explain the relevance of some of these factors and provide tips for the proper development of ELISA.
Different types of ELISA
Although ELISA can take different formats, all of them involve the same main steps-analyte capture on the surface of the microplate wells, blocking to prevent non-specific interactions, and analyte detection/quantification. In direct ELISA, the analyte is bound to the wells of a microplate and detected with a target-specific reporter antibody, while indirect ELISA uses a labeled secondary antibody for detection. In the sandwich ELISA process, the plate-bound antibody is used to capture the analyte, and the detection can be direct or indirect. Any of these ELISA methods can be converted into a competitive assay, thereby competing with the reference for binding before the target analyte is detected. As shown in Figure 2.
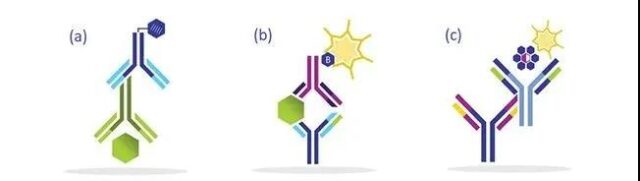
Figure 2: The working principle of the three ELISAs. (a) Indirect ELISA: involves the interaction of a fixed antigen with a sample containing a primary antibody and a labeled secondary antibody that recognizes the primary antibody. (B) Sandwich ELISA: involving immobilized capture antibody and biotin-labeled detection antibody, “sandwiched” on the bound target protein. (c) Competitive ELISA (EIA): Biotinylated peptides are added to the samples and standards, where the target protein and the labeled peptide compete with the capture antibody to interact.
Sample Preparation
ELISA can analyze a wide range of samples, including tissue culture supernatants, cell or tissue lysates, and body fluids (such as serum, plasma, or urine). In addition to the analytes of interest, many of them contain complex mixtures of biomolecules, which can cause unwanted background signals or target degradation. It is recommended that the conditioned medium sample contains no serum; if this is not possible, an uncultured medium blank should always be used as a control to evaluate the baseline signal. The buffer used to prepare cell or tissue lysates should contain protease and phosphatase inhibitors, and the use of strong denaturing detergents, sodium azide or high concentrations (>10mM) of reducing agents should be avoided. For plasma and serum samples, whole blood should be centrifuged at the time of collection, then aliquoted and stored at -80°C until use.
Choose the right buffers and reagents
Care must be taken when selecting buffers for coating, blocking, diluting antibody reagents, and washing the wells of the microplate. The function of the coating buffer is to promote the adsorption of the microplate surface and stabilize the bound biomolecules, and it must be free of protein; although PBS or TBS are viable alternatives, 0.2M carbonate/pH>9 is usually used. Bicarbonates. The blocking buffer can prevent non-specific binding, including bovine serum albumin (BSA) and various serums; BSA should be free of IgG and protease, and the serum should be from the same host species as the labeled antibody. Antibodies are usually diluted in blocking buffer, although the concentration of the blocking agent is lower. Tris buffered saline (TBS) and phosphate buffered saline (PBS) containing 0.05% (v/v) Tween-20 are widely used for washing.
Match antibody pairs and determine the optimal concentration
Sandwich ELISA is the most popular form of ELISA because it enhances specificity by using two different antibodies to recognize the target analyte. However, in order to prevent competition for target binding, it is important for each antibody to recognize a different epitope. Some antibody manufacturers provide matched antibody pairs for ELISA. If these are not available, the antibody data sheet may provide epitope mapping. Optimizing antibody concentration is critical and can be achieved using a checkerboard configuration; by titrating one analyte-specific antibody on a microtiter plate and then titrating the other antibody (while keeping all other variables constant), the maximum yield can be determined The condition of the signal-to-noise ratio.
Standard curve-determine the curve range and sensitivity
The standard curve is indispensable for quantifying the amount of target analyte present, and should be included on each test plate, ideally with repeated runs for each concentration. The curve range can be established by adding a high-concentration standard to a microplate well and titrating down to define the upper and lower detection limits; then it can be generated on each test plate by selecting the appropriate starting concentration and dilution factor standard curve line. When determining the curve range and actively testing samples, the buffer used to dilute the standard should closely match the buffer of the sample to avoid introducing artifacts.
Troubleshooting
The more time invested in optimizing the ELISA, the less likely it is to make mistakes. However, when they do, several common problems are easily solved. High background can usually be overcome by using a more concentrated blocking solution, extending the incubation time for blocking, or increasing the number/duration of washing steps. Different coating buffers or higher concentrations of analyte-specific antibodies can be used to enhance weak signals. Resolving edge effects or plate drift may only involve ensuring that the plate is tightly sealed between the addition of reagents to prevent evaporation. More troubleshooting tips for running a conventional ELISA can be found here.
How to perfect ELISA?
Choose the ELISA method that best suits your needs
ELISA can be configured in a variety of ways, all of which involve fixing the antigen to the surface of the microplate before blocking, detecting it with antibodies, and measuring the resulting signal. Although it is possible to directly immobilize antigens (a method commonly used to monitor antibodies in body fluids), sandwich ELISA is usually the first choice—especially for complex samples—because it provides greater specificity.
The ELISA method largely depends on the purpose of the experiment and the available reagents. When ELISA is set internally, the conjugated primary antibody can be used to shorten the experimental process, but in the case of low target abundance, the use of conjugated secondary antibody for indirect detection can provide valuable signal amplification. In many cases, researchers prefer to use off-the-shelf ELISA kits because they are fully optimized to save time, money, and sample materials.
Take steps to resolve background issues
Robust washing technology is one of the most effective ways to minimize the background signal in ELISA. Optimizing the number, duration and volume of washing steps, as well as the detergent concentration of the washing buffer, is essential to ensure the removal of any unbound reagents that may affect the results. In order to obtain more consistent ELISA data, an automatic plate washer may be a wise choice. Many modern instruments are equipped with functions such as programmable oscillation to eliminate air bubbles and precise pipetting to almost completely eliminate residual wash buffer.
Other methods of minimizing background in ELISA include reducing the concentration of the primary and/or secondary antibody, changing the composition of the blocking solution or the length of the blocking step, and reducing the incubation temperature. It is also a good idea to include a reference wavelength measurement to identify non-specific background noise.
Pay attention to the standard curve
The standard curve is essential for obtaining quantitative ELISA results because it is used to calculate the concentration of unknown samples. For this reason, the reference material used to produce it must be of high quality and in sufficient quantity to achieve repeatable performance throughout the life cycle of the analysis. If the reference material changes (for example, a new batch number is received), the performance should always be compared with the existing material.
ELISA data is usually expressed in terms of the concentration of the analyte relative to the measured reading, which can be colorimetric, fluorescent or chemiluminescence. Using more points within the standard curve range (ideally 8 or more) provides the flexibility to eliminate outliers and better define the curve within the signal response range to improve the fit. For curve analysis, linear fitting is usually suitable for sandwich ELISA, but the 4-parameter logistic (4PL) model can better fit the very low or high end of the measured concentration range (Figure 3). The correctness of the fitting can be verified by anti-fitting.
image
Figure 3. The same ELISA calibration curve with 2 different trend lines: linear (left) and 4-parameter regression model (right).
Ensure repeatability and accuracy
Reproducibility and accuracy are essential for any immunoassay, but when quantitative ELISA data is used to drive critical decisions (such as whether to further test potential drug candidates), the requirements can be particularly stringent.
Protective measures include increasing the number of repetitions (for example, three repetitions instead of repeated tests), not only in each experimental run, but also on each plate in the same run to merge the standard curve.
To additionally ensure reproducibility between tests, adding another internal control to supplement the standard curve can provide greater confidence in the results. As a general rule, the coefficient of variation (CV) for repeated readings should not exceed 20%.
In the case of sandwich ELISA, another good practice is to choose a plate pre-coated with capture antibody instead of an uncoated plate (development kit), as this can reduce signal variability between wells. Off-the-shelf ELISA kits can provide significant advantages here because they are fully verified and guaranteed by the manufacturer when they are provided.
How to improve ELISA performance?
Choose the ELISA that meets your needs
Although the early ELISA used immobilized antigen directly bound to the surface and detected it with a single antibody, the technology has been developed to use antibodies as capture and detection reagents. Both direct ELISA and sandwich ELISA can play a role. Direct ELISA is very suitable for detecting and measuring the concentration of small molecules because it only needs one antibody binding site, while sandwich ELISA is more suitable for larger molecules, such as proteins, because it requires two All epitopes are available at the same time. Regardless of the format chosen, it is important to confirm that the assay is sensitive enough to detect your target of interest, preferably at the baseline level of your control sample.
Even with many competing formats, sandwich ELISA is still considered the gold standard for quantitative measurement. The use of two specific antibodies eliminates considerable noise, and the sandwich structure facilitates easy washing steps and helps eliminate non-specific binding. The problem. These advantages make the sandwich ELISA have more experimental steps than the direct ELISA.
Pay attention to standards and controls
Many commercially available ELISA kits include reference materials used as standards or controls. By adding reference materials to various matrices, developers of ELISA kits can confirm that these will not cause adverse effects. However, because not every model system can be tested, researchers should check the standard provided in their own system to ensure that it does not affect the control signal.
Using reference materials calibrated in accordance with World Health Organization (WHO) standards, researchers can compare the performance of ELISA kits between laboratories and different regions. With the increasing awareness of the reproducibility crisis, the use of international standards is to ensure that An effective method of consistency. It also provides a way to compare performance with other commercial kits that meet the same WHO standards.
Follow the agreement
ELISA kit manufacturers have made a lot of optimizations to provide researchers with sensitive and reliable detection, which includes testing different antibodies, buffers and incubation conditions, as well as monitoring product stability, reproducibility, cross-reactivity and intra-batch/batch Between accuracy. The result information is summarized in the accompanying manual, as well as detailed protocols designed to ensure the success of the experiment.
Strictly following the protocol is the basis of ELISA data quality. Researchers should be consistent with the timing of various detection steps, the order in which materials are added to the plate, and the way the plate is washed and developed. He also recommends using a multi-channel pipette to increase the speed, pre-wetting the pipette tip, ensuring that all material is discharged from the tip, and changing the tip frequently to avoid cross-contamination. Material residues can be an important source of error in ELISA, which can be prevented by using disposable tips and ensuring that pipettes and automated instruments (such as plate washers) are regularly monitored to verify that they are operating as expected.
Learn about antibodies
Whether you develop your own ELISA in-house or use a kit, the detection performance depends on the quality of the antibody. Researchers developing their own detection methods should realize that antibodies validated in other applications may not be the best choice for ELISA. In addition, in order to ensure accurate ELISA results, capture antibodies and detection antibodies need to be validated together in pairs.
Consider using automation
Automation is widely recognized for its ability to improve detection reproducibility, eliminating many potential sources of variation to provide more reliable results. Researchers simply add samples and buffers to the kit, introduce them into the instrument, and walk away to avoid the pitfalls of pipetting errors, material residues, or user differences. When ELISA is performed manually, all of these will affect the result.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



