CAR-T cell therapy for solid tumors: Key lessons learned
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
CAR-T cell therapy for solid tumors: Key lessons learned
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
CAR-T cell therapy for solid tumors: Key lessons learned
Chimeric antigen receptor (CAR) T-cell therapy is an adoptive cell therapy in which autologous T cells are engineered to express CARs to specifically kill tumor cells.
Autologous CD19-targeted CAR-T cells have been approved by most major regulatory agencies, including the U.S. Food and Drug Administration (FDA), the European Medicines Agency (EMA), and the China National Medical Products Administration (NMPA).
These CAR-T cells have revolutionized clinical practice, demonstrating potent anti-tumor activity.
However, developing clinically useful CAR-T cells for solid tumor patients still faces significant challenges, largely unknown. There are many possible reasons, including:
(1) insufficiently specific target antigens;
(2) inadequate delivery;
(3) short persistence;
(4) loss of effector function;
(5) tumor antigen heterogeneity.
Therefore, it is necessary to draw lessons from past CAR-T preclinical and clinical studies, analyze the elements and challenges of CAR-T cell technology, and consider factors from basic, clinical, and practical aspects, taking countermeasures to make CAR-T technology an affordable treatment modality.
Understanding the reasons for the failure of CAR-T cells in solid tumors
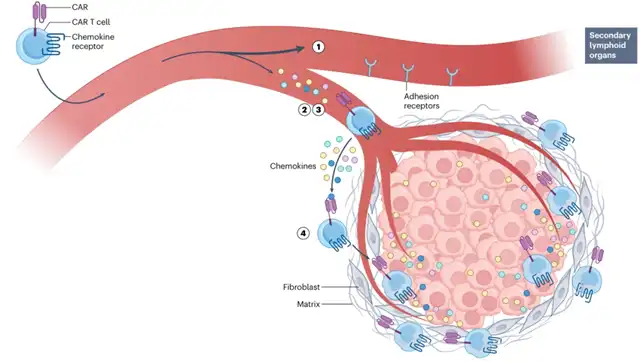
Inadequate delivery
Two fundamental questions that need to be addressed in CAR T cell research and development are where T cells go after intravenous injection and their effectiveness in entering tumors.
Preclinical data from models indicate that few injected CAR-T cells initially enter tumors, with mouse T cells injected into syngeneic mice persisting for only a few weeks. Clinical trial data in humans suggest that CAR-T cells initially accumulate in the lungs and secondary lymphoid organs, then inefficiently migrate to tumors over the next 24-48 hours.
The entry of CAR-T cells into tumors first requires recognition of chemokines secreted and bound to the surface of endothelial cells, which are predominantly located in the tumor stroma rather than the tumor cell-rich area. This initial endothelial recognition is followed by selectin-mediated rolling adhesion, then firm adhesion by integrins. Under the drive of chemokines (particularly CXCL9, CXCL10, CXCL11, and CCL5), T cells then migrate into the tumor stroma. Here, perivascular cells, extracellular matrix proteins, and stromal cells (mainly fibroblasts) form a barrier. Some T cells migrate through the stroma, with fewer T cells ultimately entering tumor cell-rich areas under the guidance of tumor-derived chemokines, where they can kill tumor cells, a process requiring binding to ICAM1 on tumor cells. This process is highly inefficient, resulting in few T cells successfully interacting with tumor cells. There are many reasons for this lack of efficiency, including chemokine-CCR mismatch, adhesive receptor defects, and the presence of extracellular matrix acting as a barrier.
Another important but underestimated issue may be the “misdirection” of CAR-T cells towards lymphoid tissues and away from solid tumors. To date, the manufacture of CAR T cells has been largely guided by data from trials testing CD19-targeted products in patients with B-cell leukemias or lymphomas, which emphasize the importance of lymph node and/or bone marrow trafficking, expansion of anti-tumor activity, and persistence. T cells with high levels of CCR7 and CD62L expression are known to preferentially traffic to lymph nodes or bone marrow. Therefore, most current protocols aim to generate CAR-T cells primarily with a central memory cell (CD62L high and CCR7 high CD45RO+) phenotype rather than an effector memory cell (CD62L low and CCR7 low CD45RO++) phenotype, which is typically disadvantageous for trafficking to tumors.
Furthermore, two additional considerations should be taken into account. Firstly, cryopreservation may affect CD62L expression, thus affecting trafficking. Secondly, CD62L is thought to promote anti-tumor activity through other mechanisms, specifically, CD62L may guide T cells to high endothelial venules within tertiary lymphoid structures present in certain solid tumors. These lymphoid structures may facilitate T cell infiltration and improve anti-tumor immune responses.
Short persistence
In clinical trials involving solid tumor patients, with regard to the persistence of CAR-T cells, using PCR or flow cytometry detection, nearly every trial conducted so far has shown that CAR-T cells exist only in blood samples. The range of CAR T cell DNA transcripts in blood is 103 to 104 copies per microgram of DNA, and CAR-T cells are detected only within about a month after infusion, with the peak usually occurring at 10-14 days. In contrast, most successful trials testing CD19-targeted CAR-T cells in leukemia patients have found large numbers of CAR-T cells in the blood, typically 105-106 copies per microgram of DNA, with durations ranging from months to years.
In summary, preclinical and clinical study results indicate that the efficiency of CAR-T cell transport to tumors is very low after intravenous injection, and most of this transport may occur shortly after injection. Limited data from human studies suggest that the few CAR-T cells that enter solid tumors have limited persistence and do not proliferate widely.
Loss of effector function
Preclinical studies of CAR-T cells consistently find that their cytotoxic activity is initially high but gradually decreases over time. Genomic and epigenomic changes are associated with this low-functioning state. The reasons for this progressive loss of function are not fully understood. It may be related to various factors present in the TME, including low pH, hypoxia, nutrient deficiencies due to low levels of key amino acids and glucose, high levels of reactive oxygen species (ROS), the presence of immunosuppressive mediators (such as TGF-β, PGE2, adenosine, and IL-10), and interactions with bone marrow-derived suppressor cells and CD4+ regulatory T cells. Intrinsic factors in T cells include “regulatory shutdown” mediated by immune checkpoints (such as PD-1, CTLA4, TIM3, TIGIT, and LAG3) and inhibitory intracellular signaling pathways (such as DGK, NR4A, SHP1, and cbl-b). Additionally, there are numerous epigenetic changes.
Tumor heterogeneity and antigen spreading
Unlike in B-cell malignancies or multiple myeloma, antigen expression in solid tumors is almost always lower and more heterogeneous. Therefore, the likelihood of successful treatment is low unless CAR-T cells induce some bystander or antigen-spreading effect, or target multiple antigens.
Lessons to be learned
Some solid tumor patients can now be successfully treated with adoptive T-cell transfer, with the most notable success being TIL therapy in melanoma patients. TILs have also shown promising results in non-small cell lung cancer patients. What can we learn from these successes to achieve greater success with CAR-T cells in solid tumors?
T cells optimized for lymph node trafficking may not be the best cells for targeting solid tumors
Compared to effector cells or effector memory cells preferentially trafficked to peripheral sites, T cells with high expression of CD62L and CCR7 on naive or central memory T cells can be preferentially trafficked to secondary lymph nodes and bone marrow. When used to develop CAR-T cells targeting solid tumors, cells with these phenotypes may be suboptimal. Solid tumor-targeted CAR-T cells are designed to target intact antigens expressed on the surface
of solid tumor cells. Therefore, when these cells enter the bone marrow or lymph nodes, they will not encounter their antigens, and thus will not be activated to proliferate or differentiate into effector cells, unless the tumor has metastasized to these locations. Data show that in solid tumor models, effector CAR-T cells are more effective than memory T cells. Therefore, the key lesson that solid tumors need to avoid is the standard approach of generating CAR-T cells designed to optimize lymph node trafficking.
DC interactions may be important
One important difference between TILs or TCR-T cells and CAR T cells is that the former can be broadly activated by DCs, which can provide optimal costimulatory signals. This costimulation can occur in lymph nodes and bone marrow. Finding ways to harness the activating ability of DCs to enhance CAR-T cell proliferation and persistence is an experience we should try to draw from trials involving TILs.
Long-term persistence may not be necessary
It is generally believed that the success of CAR-T cell therapy is closely related to the persistence of CAR-T cells. However, the importance of persistence has not been confirmed in clinical trials testing CAR-T cells in solid tumor patients for various reasons. Additionally, data from solid tumor models suggest that the persistence of CAR-T cells is associated with an increase in the level of T cell dysfunction.
One approach to addressing this challenge is to modify CAR-T cells to make them more persistent and less prone to dysfunction. Over the past few years, some encouraging successes have been achieved in solid tumor patients using this approach. However, another approach that needs to be considered is not focusing on persistence but using a strategy of repeated dosing to administer fresh CAR-T cells. Over time, the injected CAR-T cells will be exhausted, and they can be replaced by new doses of highly active CAR-T cells. It is necessary to reduce the immunogenicity of the CAR component to avoid rejection of the cells given multiple times.
Targeting multiple tumor antigens is important
In addition to differences in activation mechanisms, an important difference between TILs and CAR-T cells is that the former are inherently polyclonal, and therefore may target more than one antigen. Any successful CAR-T cell therapy for solid tumors will need to target multiple tumor-specific antigens, ideally triggering some bystander effect or inducing antigen spreading to the endogenous immune system. For example, designing CAR-T cells that secrete FLT3 ligands to recruit DCs, along with using DC activation with a 4-1BB agonist, to promote endogenous T cell responses and enhance anti-tumor activity.
How to better develop CAR-T
To address these challenges, it may be considered to move forward through two different strategies by optimizing CAR-T cell therapy to achieve greater success in solid tumor patients. Strategy 1 is the current direction in the field, based on the traditional “memory cell” model developed for patients with hematologic malignancies. Strategy 2 employs a different “short-lived effector cell” model.
Strategy 1: Memory cell model
The goal of this approach is to maximize the transport and persistence of CAR-T cells while minimizing the degree of CAR T dysfunction. As part of the strategy, high-dose lymph node clearance chemotherapy is used to enhance engraftment. The persistence and low function of CAR-T cells have been addressed by introducing specific genetic changes (e.g., through CRISPR) to suppress possible inhibitory factors, allowing T cells to “rest,” thereby preventing chronic stimulation, or directly by using armed CAR-T cells capable of secreting cytokines or other proteins to alter the TME.
A major challenge associated with this strategy is the need to introduce multiple genetic changes. Over the past few years, many studies have been able to knock out one or more genes using standard CRISPR techniques, and many more advanced CRISPR techniques have been developed that can efficiently edit genes with very low off-target mutation rates.
The development of allogeneic T cells derived from induced pluripotent stem cells (iPSCs) can also provide an alternative approach to improve the transport and persistence of CAR-T cells by allowing multiple genetic changes to be made. iPSCs will first be modified using various CRISPR guides to knock down selected genes, making the cells less immunogenic, then using guides for selected key inhibitors to knock down T cell function. After selecting clones with all expected changes, additional genes can be added to enhance transport or function.
Strategy 2: Short-lived effector cell model
Although not well-studied, some data support the superiority of cells with a shorter lifespan that more closely resemble effector T cells in solid tumor mouse models. For example, it has been found in studies of mesothelin-targeted CAR-T cells that cells with a CD28-CD3ζ intracellular domain structure (more like effectors) have better anti-tumor activity than cells with a 4-1BB-CD3z intracellular domain structure.
Given that persistence is not the goal, strategy 2 may also use mRNA-transduced CAR T cells. One obvious advantage of this approach is that there is no limit to the size of the transgene, and multiple mRNA can be introduced and expressed simultaneously through electroporation, allowing for the introduction of multiple genetic variations.
However, a potential limitation of using mRNA-transduced CAR T cells prepared in this way is the cost issue of administering multiple doses of CAR-T cells over time, compared to the “one-time” approach in strategy 1. Although patients would need to be re-infused, this is similar to the administration of chemotherapy or immune checkpoint inhibitors, typically administered every 4-6 weeks, sometimes for many years.
Conclusion: CAR-T cell therapy for solid tumors: Key lessons learned
Addressing the numerous challenges faced by CAR-T cell therapy in the treatment of solid tumors is a daunting task. Current treatments are largely ineffective for solid tumor patients, and the reasons for this are not fully understood. Obtaining more and better information on the transport, persistence, and function of CAR-T cells in patients undergoing treatment is crucial to addressing these aspects of the problem.
A large number of obstacles to treatment success may require targeting multiple immune evasion mechanisms. These strategies will include optimizing CAR engineering, optimizing conditions during ex vivo T cell expansion to promote the appearance of specific T cell subtypes, and manipulating the host to change the TME or stimulate natural T cells, all of which may require using all of these methods to achieve optimal results.
CAR-T cell therapy for solid tumors: Key lessons learned
References:
1.CAR T cell therapy for patients with solid tumours: key lessons to learn and unlearn. Nat Rev Clin Oncol.2023 Oct 30
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



