Why did FDA approve Aducanumab for AD even most experts objected?
- A Single US$2.15-Million Injection to Block 90% of Cancer Cell Formation
- WIV: Prevention of New Disease X and Investigation of the Origin of COVID-19
- Why Botulinum Toxin Reigns as One of the Deadliest Poisons?
- FDA Approves Pfizer’s One-Time Gene Therapy for Hemophilia B: $3.5 Million per Dose
- Aspirin: Study Finds Greater Benefits for These Colorectal Cancer Patients
- Cancer Can Occur Without Genetic Mutations?
Why did FDA approve Aducanumab for AD even most experts objected?
Why did FDA approve Aducanumab for AD even most experts objected? Most experts objected, and the three judges resigned. Why did FDA approve this new drug for Alzheimer’s disease?
In the current wave of accelerated approval of aducanumab, it is not difficult to find that the advantages and disadvantages of FDA accelerated approval program are extremely prominent…
On June 7, the U.S. Food and Drug Administration (FDA) accelerated the approval of aducanumab as a disease-modifying therapy for Alzheimer’s disease.
As Alzheimer’s disease had only a few drugs that improve symptoms before it was on the market, the news immediately triggered widespread discussions in the scientific community-Alzheimer’s disease patient organizations welcomed the drug’s listing and chanted the FDA It gave patients “new hope”; on the other hand, the FDA’s advisory committee not only appeared in the awkward scene of 10 votes against and 1 abstention in the vote on whether to recommend the drug to be marketed, but even after its listing, there were 3 advisory committees. The committee member resigned due to dissatisfaction with the FDA.
Although the quarrel between the two sides continues, what we can be sure of is that the FDA broke the routine more than once when the drug was on the market, and the disease targeted by the drug was too compelling, which led to the current series of events. Reviewing the listing process of aducanumab will undoubtedly bring some enlightenment to companies seeking the listing of new drugs.
Doubt 1: Is the alternative end point reliable?
According to the announcement issued by the FDA and Biogen’s press release, it is not difficult to find that the FDA is accelerating the approval of aducanumab on the market based on “aducanumab is beneficial to clearing β-amyloid plaques (Aβ) in the brains of patients with Alzheimer’s disease.” of. This is actually the place where the drug has been most questioned-because there is currently no direct evidence that the elimination of Aβ can improve the course of human Alzheimer’s disease. It can even be said that Aβ is the cause of Alzheimer’s disease. It is not strong yet (some scholars believe that neurofibrillary tangles caused by misfolding of Tau protein are the real cause of Alzheimer’s disease). Questioners believe that given that aducanumab is extremely expensive (US$56,000 per year), will the use of “so sloppy” alternative endpoints to approve the drug will really benefit patients from the disease?
Let us first take a look at the FDA’s “Accelerated Approval Program” (Accelerated Approval Program).

FDA’s accelerated approval project Picture: FDA
The accelerated approval project was established by the FDA in 2002. The purpose of the project is that taking into account the characteristics of certain diseases, routine clinical studies of drugs may seriously delay the drug’s marketing process; at the same time, for certain diseases, the fatality rate is extremely high and the disease course is short. It is very important to convert clinical trial drugs into available therapies in the early stage. Therefore, the accelerated approval program can temporarily approve the drug based on the alternative endpoint, while waiting for the large clinical study data to determine the drug’s benefit, and then decide to officially approve the drug or withdraw it Marketing authorization. According to the information disclosed by the FDA, between 2011 and 2012 alone, 12% of the FDA’s new drugs were approved through the accelerated approval program.
For pharmaceutical companies, the “alternative end point” is not something you can choose if you want to. In general, “surrogate endpoints” can be divided into three categories:
1. The surrogate endpoint is directly related to disease remission: if the improvement of the surrogate endpoint can directly achieve the goals of disease relief and reversal of the disease course, then the company only needs to provide data on the drug to improve the surrogate endpoint before it can be listed, and there is no need to apply for accelerated approval in advance. The typical representatives in this regard are all kinds of drugs for the treatment of hypertension. According to the FDA’s relevant guidelines, hypertension drugs only need to prove that they can significantly reduce blood pressure, and the drug can be marked on the label “lowering blood pressure can reduce fatal or non-fatal cardiovascular events, mainly the risk of stroke and myocardial infarction.” .
2. Surrogate endpoints are powerful markers of disease prognosis: For some slow-course diseases, clinical trials may take extremely long, but if there are known prognostic biochemical markers for such diseases, then companies can first prove that drugs can improve The level of prognostic biochemical markers will be accelerated after approval for larger scale studies. A typical case in this regard is Obeticholic Acid, which is used by Intercept to treat primary biliary cholangitis. This drug can effectively reduce the alkaline phosphatase level of patients in clinical studies. A large number of previous studies have It is confirmed that the higher the alkaline phosphatase level of patients with this disease, the worse the prognosis.
3. The surrogate endpoint only temporarily reflects the effectiveness of the drug, but the disease lacks treatment methods: this “surrogate endpoint” is usually used in the marketing application of anti-tumor drugs. Some tumors progress very fast, but there may be a lack of test patients for large-scale trials. For the treatment of such tumors, the overall tumor remission rate (ORR, refers to the proportion of patients in the study group who achieved remission in the overall patient), remission Indicators such as duration (DoR, patients after taking drugs to relieve the disease, the time from remission to progress again) and other indicators can be used as “surrogate endpoints” to allow drugs to serve patients who lack hope as soon as possible.
In contrast to the approved aducanumab this time, the surrogate end point chosen seems to be a “powerful marker for disease prognosis”, but as mentioned above, it is doubtful whether Aβ is a prognostic marker for disease, and whether there is some choice for this end point. Too sloppy?
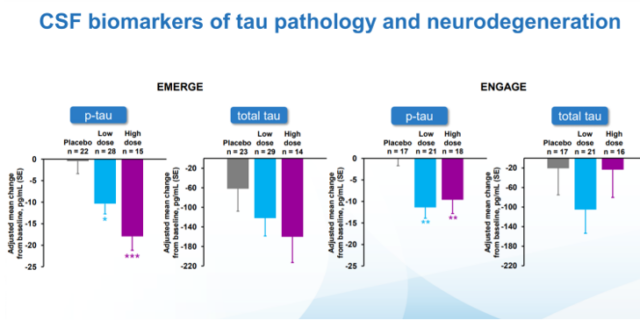
Aducanumab has used a variety of biomarkers as research endpoints in clinical studies, but their significance is not yet clear. Picture: Biogen
Doubt 2: Contradictory clinical research
If a drug is to be marketed, clinical studies must be conducted to prove its effectiveness against the disease. In an ideal situation, if the drug is successful in clinical trials, the drug regulatory authority will review the relevant data and finally decide whether to approve the drug. However, not all clinical studies of drugs will achieve ideal results. For clinical studies of drugs with poor results, the FDA will pay special attention to it, and even often reject these drugs with conflicting research results on the grounds of “insufficient clinical data”. .
A classic example of this is the antiplatelet drug cangrelor used in coronary interventional surgery. After 10 years of development of the drug, three large-scale phase III studies have been conducted to explore its effectiveness, but two of the studies failed, and the third study was also cited by the FDA as “inadequate clinical study design and data analysis” as the reason Rejected. It may be because the research and development company The Medicines Company spent a lot of research and development funds on the drug, and subsequently conducted a second analysis of the CHAMPION PHOENIX study, which was declared successful by Cangrelor, and finally “impressed” the FDA and successfully marketed the drug.
This case tells us that a problematic clinical study can have a devastating impact on the marketing of the drug.
In the process of aducanumab’s listing, questionable clinical studies did not seem to have any impact on drug review. As we all know, Biogen has conducted two clinical studies of EMERGE and ENGAGE for aducanumab. Although both studies have produced positive results, their data are contradictory: in the EMERGE study, observation of patients who applied high-dose aducanumab The score of CDR-SB on the dementia rating scale improved (22% reduction compared with the placebo component, p=0.0120), but in the ENGAGE study, the same dose of aducanumab had little effect on the patients’ dementia symptoms, and even showed The tendency to worsen the symptoms of dementia (2% higher than the number of placebo components, p=0.8330).
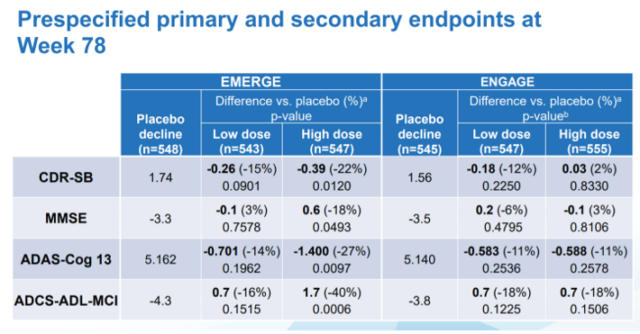
List of clinical results data of aducanumab announced by Biogen. Picture: Biogen
What is even more incredible is that this data tends to appear in most dementia scores:
Concise Mental State Scale (MMSE) score: In the EMERGE study, the number of components compared to the placebo was reduced by 18% (p=0.0493), and the number of components in the ENGAGE study was increased by 3% (p=0.8106, not statistically significant) Sex);
Alzheimer’s Disease Evaluation Scale-Cognitive Score (ADAS-Cog12) score: The number of components in the EMERGE study was reduced by 27% compared to placebo (p=0.0097), and the number of components in the ENGAGE study was reduced by 11% compared to the placebo. (P=0.2578, not statistically significant);
Alzheimer’s activity of daily living scale score for mild cognitive impairment: 40% reduction in the number of placebo components in the EMERGE study (p=0.0006), and 18% reduction in the number of placebo components in the ENGAGE study (P=0.1506, not statistically significant).
Judging from these data, the improvement of the symptoms of Alzheimer’s disease by aducanumab is very poor in different studies. According to the FDA’s practice, the drug would have been required to do the third phase III study, or go further. After collecting the data of the two studies, the analysis will be carried out. However, it may be due to the lack of breakthroughs in the field of Alzheimer’s disease for too long. The FDA still took the risk to approve the application of aducanumab, which can be said to be very unreasonable.
Doubt 3: The patient’s attitude determines the approval of the drug?
During the approval process of aducanumab, the FDA’s expert committee rejected the recommendation for approval of aducanumab for Alzheimer’s disease with an overwhelming result of 10 votes against and 1 abstention. Although the FDA will follow the advice of the expert committee in principle, this time the FDA still bypassed the expert committee and chose to approve aducanumab for the treatment of Alzheimer’s disease. Some doctors and the media accused that the FDA approval of the drug may have been affected by the lobbying of patient organizations.
According to previous interviews, the chief scientific officer of the American Alzheimer’s Disease Association once stated that the approval of the drug “at least gives Alzheimer’s disease patients some hope that they can try, rather than allowing them to gain more Waiting for years for positive results with certainty”. Under this policy, when aducanumab received accelerated approval from the FDA, the American Alzheimer’s Association has been lobbying vigorously in the media and other public places, hoping that the drug will be approved.
“Off-market” affects drug approval, and aducanumab may not be alone. Flibanserin, a drug approved by the FDA in August 2015 for the treatment of female sexual desire loss dysfunction (HSDD), has unsatisfactory results in clinical studies, and even has side effects such as hypotension and syncope. Although the drug was rejected by the FDA once in 2010, the drug was still approved by the FDA under the lobbying of the patient population. It’s just that because the drug’s comprehensive advantages are not obvious, the medical community has been publishing articles criticizing the FDA since the drug’s launch. What’s even more embarrassing is that the market for flibanserin has been extremely bleak after the launch of the new generation of drugs. Pharmaceutical sales in 2018 were only 3.48 million U.S. dollars.

Flibanserin approved in the dispute is also problematic
Discussion: will “Accelerate approval” be the bad example for controversial or immature drugs?
During the accelerated approval of aducanumab, it is not difficult for us to find that the advantages and disadvantages of the FDA accelerated approval program are extremely prominent: the program gives potential new drugs the right to “get on the bus before making up the ticket”, allowing patients to use it faster In addition to life-saving drugs, some drugs with weak clinical research data have been “speculatively exploited” and approved by the FDA.
In the current drug approval policy, there are already many special cares (such as FDA’s “Orphan Drug Qualification”, “Breakthrough Drug Qualification”, China National Medical Products Administration (NMPA) “Priority Review Procedure”, “Clinical urgent need for overseas new drug qualification” Today, the status of accelerated approval of projects is undoubtedly worthy of our rethinking.
Of course, fortunately, accelerated approval is not a “one-shot deal.” Under the premise that the drug’s later clinical research data cannot support its listing, regulatory agencies such as the FDA are eligible to revoke the accelerated approval. For example, Olaratumab, a new drug previously used for soft tissue sarcoma, was granted accelerated approval by the FDA on October 19, 2016 because of its remarkable efficacy in previous studies.
However, after the phase III clinical study of the drug failed, the FDA announced that it would suspend the use of the drug in 2019. After the NMPA also granted the drug a “priority review procedure”, it referred to foreign clinical research data during approval and made a decision on Olaratumab. “Disapproval” administrative decision.
As for the fate of this “storm core” aducanumab after the accelerated approval, we may as well wait and see.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.