The importance of fusion gene NGS multi-platform detection of NSCLC
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
The importance of fusion gene NGS multi-platform detection of NSCLC
The importance of fusion gene NGS multi-platform detection of NSCLC: the non-classical fusion gene detected by the DNA platform needs to be corrected by the RNA platform before targeted therapy.
More and more rare ALK/ROS1/RET fusion partners or complex translocation types in lung cancer have been reported and discovered, but their authenticity and the relationship with the efficacy of clinical targeted therapy need to be further verified by large samples.
Next-generation sequencing (NGS) technology can simultaneously detect multiple gene mutation types in non-small cell lung cancer (NSCLC), which can be based on DNA or RNA. In clinical testing, DNA-based NGS testing has greater advantages. Therefore, currently approved NGS testing kits are mainly DNA-based NGS platforms.
With the widespread clinical application of DNA-NGS, more and more rare ALK/ROS1/RET fusion partners or complex translocation types in lung cancer have been reported and discovered, but its authenticity and its relationship with the efficacy of clinical targeted therapy need further Large sample verification.
Our young doctoral team of the Yangtze River Delta Lung Cancer Collaboration Group has conducted in-depth exploration and analysis on this issue in conjunction with Hengte Gene, and published an editorial in the early stage. The editorial mainly commented on three aspects. The first aspect is the China Ying Jianming team. Research (JTO published a paper comparing the clinical significance of DNA-NGS and RNA-NGS detection of fusion genes) (RNA NGS detection based on Hengte’s PANO-Seq® technology) and MSKCC research both passed RNA NGS and negative non-small cells in DNA NGS detection About 10% of target gene fusions or MET skip mutations (MET-MET fusion) were detected in lung cancer samples. Among them, ROS1 fusion is the most common, possible reasons include:
(1) The intron region at the common breakpoint of ROS1 fusion is larger (31-35 intron region), which requires more probe coverage;
(2) Low GC content leads to low probe capture efficiency and low sequencing depth;
(3) A high degree of AT repetition area affects the accuracy of subsequent data comparison analysis.
Therefore, for NSCLC samples with negative DNA NGS tests, other platforms should be considered when necessary to further verify the accuracy of fusion genes, especially ROS1.
In the second aspect, for the rare fusion partners of ALK, ROS1 and RET detected by DNA NGS, 6 cases of ALK/ROS1 fusion were not detected at the RNA level through the RNA NGS detection of Hengte PANO-Seq® technology, 5 of them No significant curative effect has been obtained after receiving targeted therapy.
This rare fusion, which is only detected at the DNA level, is not effective for targeted therapy. It may be the main reason why the primary/reciprocal fusions of ALK (primary/reciprocal fusions) have been found to be less effective than single EML4-ALK fusions in targeted therapy. The importance of further functional verification at the RNA or protein level for non-classical fusions detected by DNA NGS.
Third, even if the PANO-Seq® technology RNA NGS is positive, most of the fusion partners determined by DNA NGS and PANO-Seq® technology RNA NGS are not consistent. Chromosome fragmentation may be an important reason for the differences in DNA and RNA levels of ALK, ROS1 and RET fusion partners.
It is worth noting that for the rare ALK and RET fusions detected by DNA NGS, most of the partner genes are on the same chromosome as ALK and RET, while this phenomenon does not exist in ROS1 fusions (Figure 1).
For mutual fusions, it can also be seen that 5’reverse fusions are mostly located on the same chromosome with the corresponding ALK and RET fusions, further suggesting that it may be formed by non-homologous end joining of DNA double strands after chromosome fragmentation. Most of them will be cut off during the transcription process. Therefore, for the mutual fusion of ALK and RET, PANO-Seq® technology RNA NGS only detects 3’fusions but not 5’fusions. 5’fusions at the DNA level may be “passenger fusions”. For LK/ROS1 alone /RET non-classical 3’fusion, PANO-Seq® technology RNA NGS detected mostly classic fusions.
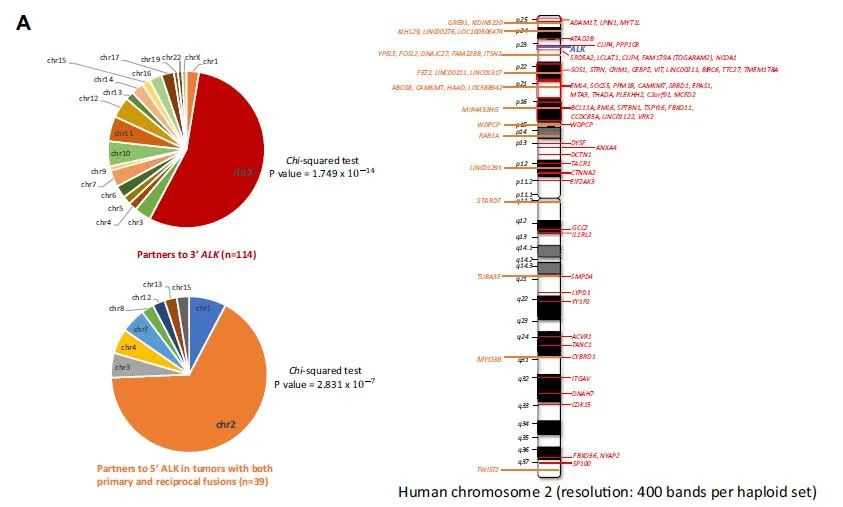
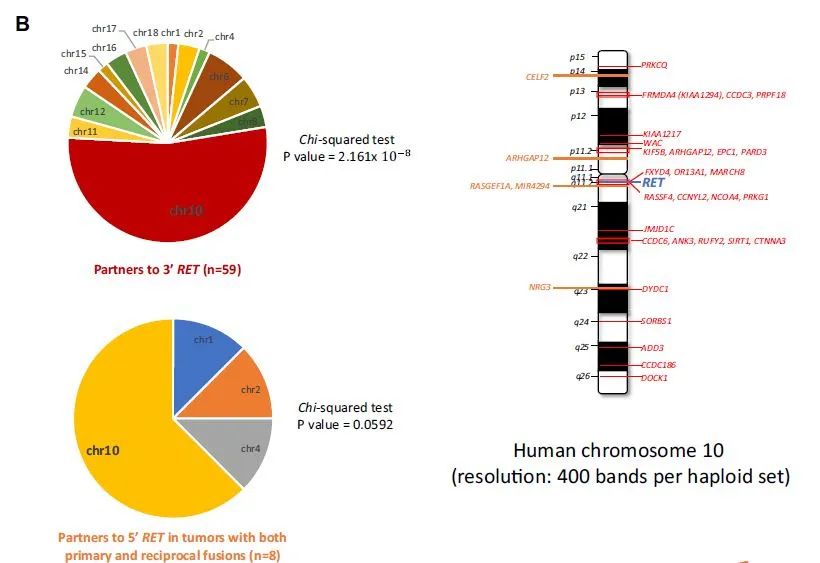
Figure 1. The rare fusion partners of ALK and RET detected by DNA NGS are mostly located on the same chromosome.
Our young doctor team of the Yangtze River Delta Lung Cancer Collaboration Group believes that while DNA NGS detection is fusion, other detection methods such as PANO-Seq® technology RNA NGS/IHC for verification can better guide subsequent clinical targeted therapy, especially It is a very common fusion partner/type, and the consensus of multi-platform detection of gene fusion in NSCLC should be further improved and promoted.
Factors of Gene Fusion Variation
A review published by MD Anderson Cancer Center in 2019 summarized the main reasons that cause fusion rearrangement and translocation, that is, the rearrangement of the chromosome layer or the wrong splicing during transcription. Rearrangement at the DNA level mainly includes six forms: intrachromosomal translocation and inter-chromosomal translocation, insertion of large fragments, deletion of large fragments, tandem duplication, inversion, or complex rearrangement caused by chromosome fragmentation (figure 2).
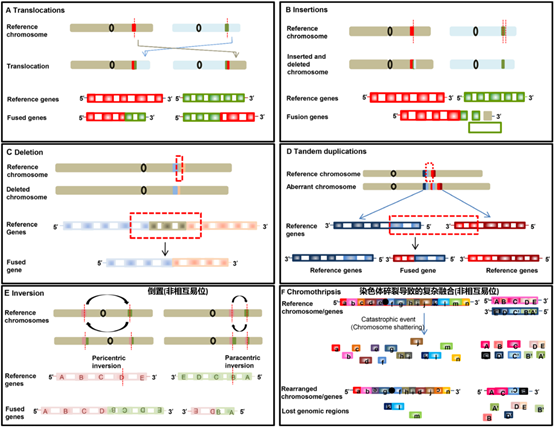
Figure 2. The 6 major factors that cause gene rearrangement/fusion at the DNA level.
At the RNA level, the “read-through event” in the transcription process may also produce fusion proteins. When RNA polymerase cannot correctly terminate the transcription of one gene end, and the transcription continues to the next gene end due to abnormal splicing, a chimera occurs. Transcripts are called “read-through events”, and fusion transcripts can be produced without involving the rearrangement of genomic material (DNA rearrangement) (Figure 3).
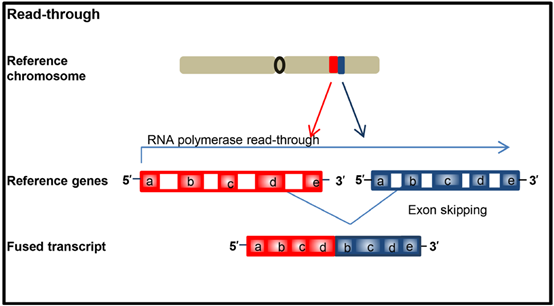
Figure 3. The rearrangement/fusion caused by the “read through” event generated during the transcription process does not involve the genomic level and cannot be reflected by DNA analysis.
In addition, RNA alternative splicing and RNA skipping (RNA Skipping, where one or some of the exons are mistakenly deleted or replaced due to incorrect splicing of the mRNA precursor) can also produce fusion repetitions. Row (Figure 4).
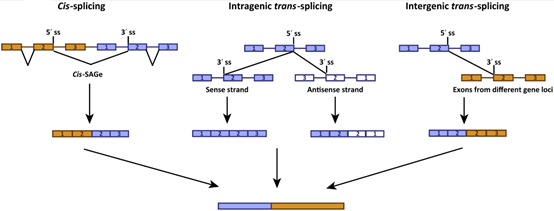
Figure 4. Alternative splicing during mRNA maturation processing mainly produces rearrangements/fusions within genes, such as skipping of exon 14 of MET in NSCLC.
Among the many factors that cause gene rearrangement/fusion, chromosome fragmentation(Chromothripsis) and chromosome weaving (Chromoplexy) are the main reasons for complex rearrangements at the DNA level. So that it is often detected at the DNA level: gene-intergenic region fusion, intergenic region-intergenic region fusion, or detected in the same sample: mutual fusion and non-mutual fusion coexist at the same time, the target gene and Very complex fusion/rearrangement events such as fusion of multiple partners at multiple splicing points (Figure 5, 6).
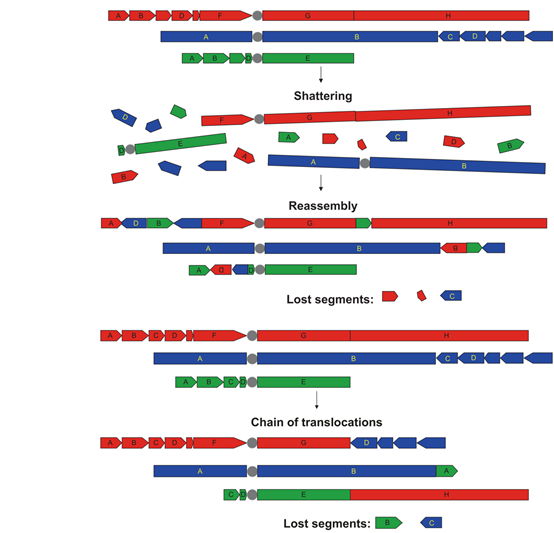
Figure 5. Chromothripsis: DNA double-strand breaks, which causes chromosomes to split into many fragments, and then under the DNA self-repair mechanism, these fragments are randomly reconnected together, causing one or more chromosomes to rearrange , Forming a mosaic chromosome. Chromosome fragmentation can cause large-scale rearrangement of the genome, which can accelerate the rearrangement of genomic DNA instead of gradually acquiring rearrangements and mutations over time.
Chromoplexy: Double-stranded DNA breaks on different chromosomes cause disordered strand rearrangement, that is, strand translocations between multiple chromosomes are involved, and fragment deletions often occur at the breakpoints.
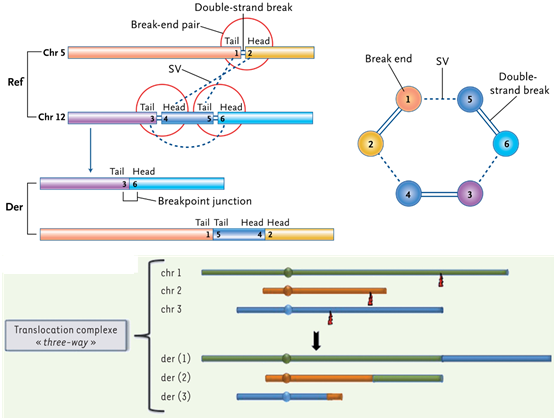
Figure 6. Schematic diagram of complex fusion mutation events caused by chromosome fragmentation and chromosome weaving on the genome.
Related studies also show that a considerable proportion of fusion mutations in tumors are caused by chromosome fragmentation and chromosome weaving.
The “Pan-Cancer Analysis of Whole Genomes Consortium” (PCAWG) study published on Nature in February 2020 showed that on average, each cancer genome carries about 4 or 5 driver mutations, which gives These cancer cells bring a selective advantage in survival, and only 5% of the tumors in this study did not find driver mutations.
The study also found that many cancers exhibit genomic disasters–complex chromosomal rearrangements: 17.8% of tumors have Chromoplexy (chromosome weaving) events and Reciprocaltranslocations (mutual translocations); 22.3% of tumors have Chromothripsis (chromosome fragmentation). ).
A study published on Cell in June 2019 showed that by analyzing the whole genome sequencing data of 138 lung adenocarcinoma samples, it was found that 74% of lung adenocarcinoma patients (Smoking-signature-lowLADCs) with low correlation with smoking (Smoking-signature-lowLADCs) The known fusion oncogenes are produced by complex genome rearrangements such as Chromothripsis (chromosome fragmentation) and Chromoplexy (chromosome weaving) (including EML4-ALK, CD74-ROS1 and KIF5B-RET, etc.).
Fusion oncogenes are produced in the early stages of tumorigenesis, and there is a long diagnosis delay. The source of 39 tumor-driven fusion genes was analyzed, of which 10 cases (26%) were produced by simple recombination, including large fragment deletion, Reciprocalinversions (mutual inversion), Reciprocaltranslocations (mutual translocation), etc.; 29 cases (74%) were caused by complex (The average number of rearrangement breakpoints is up to 20), of which 5 cases are Chromoplexy with multiple breakpoints distributed on more than two chromosomes, 3 cases are typical Chromothripsis (within a single chromosome), and 9 cases In order to involve 1-2 chromosomes with the characteristics of Chromoplexy and Chromothripsis acting together (clustered breakpoints, and the copy number is basically balanced), 5 cases are Chromothripsis among multiple chromosomes (Figure 7).
It can be seen that the formation of fusion oncogenes in LADCs is caused by multiple types of complex recombination, which may be the initial event of malignant canceration of normal respiratory epithelial cells, especially for non-smokers.
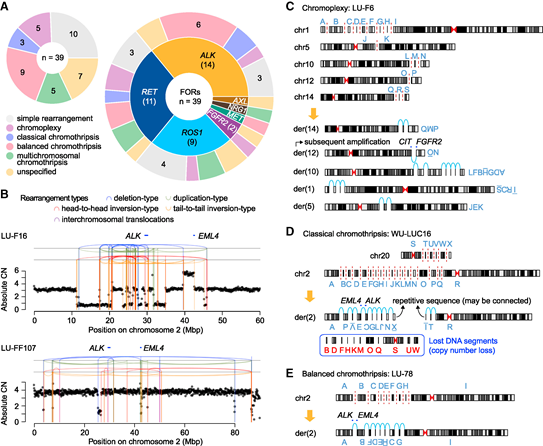
Figure 7. Complex genome rearrangement produces fusion oncogenes:
- (A) Genome rearrangement pattern of fusion oncogenes in lung adenocarcinoma.
- (B) An example of Chromothripsis that produces EML4-ALK fusion oncogene. In case LU-F16, many DNA fragments were lost during the recombination process after chromosome fragmentation, and the final footprint showed a typical copy number imbalance; in contrast, case LU-FF107 showed a copy number balance pattern. Rearrangements are described as vertical lines and connecting arcs, and their colors indicate different types of rearrangements. CN represents the number of copies. (CE) Examples of chromosome weaving and chromosome fragmentation of the driver fusion oncogene that produce lung adenocarcinoma:
- (C) classic chromosome fragmentation,
- (D) balanced chromosome fragmentation,
- (E) driver fusion in lung adenocarcinoma Oncogenes, capital letters indicate DNA fragments and their orientation corresponding to their rearrangement configuration in the derived chromosomes. The red dashed line indicates a DNA double-strand break, and the light blue arc indicates the connection break point.
So, can these complex fusion events occurring at the DNA level form fusion products at the RNA level? What is the form of the final product? Can the information about the fusion of the target gene and the intergenic region detected at the DNA level accurately predict the true RNA product? Does the complex fusion event detected at the DNA level in the clinic actually participate in the translation and transcription process of the fusion mutation and finally express a functional fusion protein? Related phenomena have attracted more and more attention from clinical experts. Accurately predicting the true fusion mutation pattern on the transcriptome, and identifying the fusion mutation that can ultimately be translated into the fusion protein that drives tumorigenesis has very practical clinical significance and urgently need to be studied in depth.
RNA NGS is a better technical solution for detecting gene rearrangement/fusion
It is true that DNA-NGS can find information about regions that RNA-NGS cannot find, such as intergenic regions (IGR) and non-coding regions (UTR). However, a large number of studies have confirmed that the RNA-NGS method is particularly efficient for discovering gene fusion and its mechanism, and can provide direct evidence to support whether the observed fusion actually occurs, and at the same time provide evidence for whether the fusion gene is expressed.
RNA-NGS detection can more intuitively analyze the true fusion on the transcriptome and judge its biological function; further subdivide these patients who have less survival benefit from targeted therapy, and identify those patients who cannot get the corresponding TKI Benefit from the treatment, so as to avoid ineffective treatment.
Medical experts believe that the methods of RNA-NGS and WTS are particularly effective for discovering gene fusion and its mechanism. The reason is that mRNA-Seq can provide direct evidence to support whether the observed fusion has occurred and at the same time provide evidence for whether the fusion gene is expressed.
DNA-NGS and WES can find information about regions that mRNA-Seq cannot find, such as the fusion/rearrangement of intergenic regions and UTR. Therefore, the comprehensive use of multi-platform detection can fully understand the fusion and mutation of the complex tumor genome.
(source:internet, reference only)
Disclaimer of medicaltrend.org



