Progress of EGFR allosteric inhibitors for non-small cell lung cancer
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Progress of EGFR allosteric inhibitors for non-small cell lung cancer
Progress of EGFR allosteric inhibitors for non-small cell lung cancer. Lung cancer is the second largest cancer in the world, with a high prevalence and fatality rate, accounting for about one-third of the total number of cancer-related deaths each year.
Non-small cell lung cancer (NSCLC) is the main subtype of lung cancer, accounting for 85% of all lung cancer cases.
Epidermal growth factor receptor (EGFR) belongs to the ErbB or HER/ErbB family, and is a cell surface receptor tyrosine kinase (RTK).
It contains a cysteine-rich extracellular ligand binding domain, an intracellular domain containing a tyrosine kinase (TK) site, and a C-terminal tail containing an autophosphorylation site.
EGFR plays an important role in a variety of cell signaling pathways for cell survival and proliferation, migration, angiogenesis and cell death.
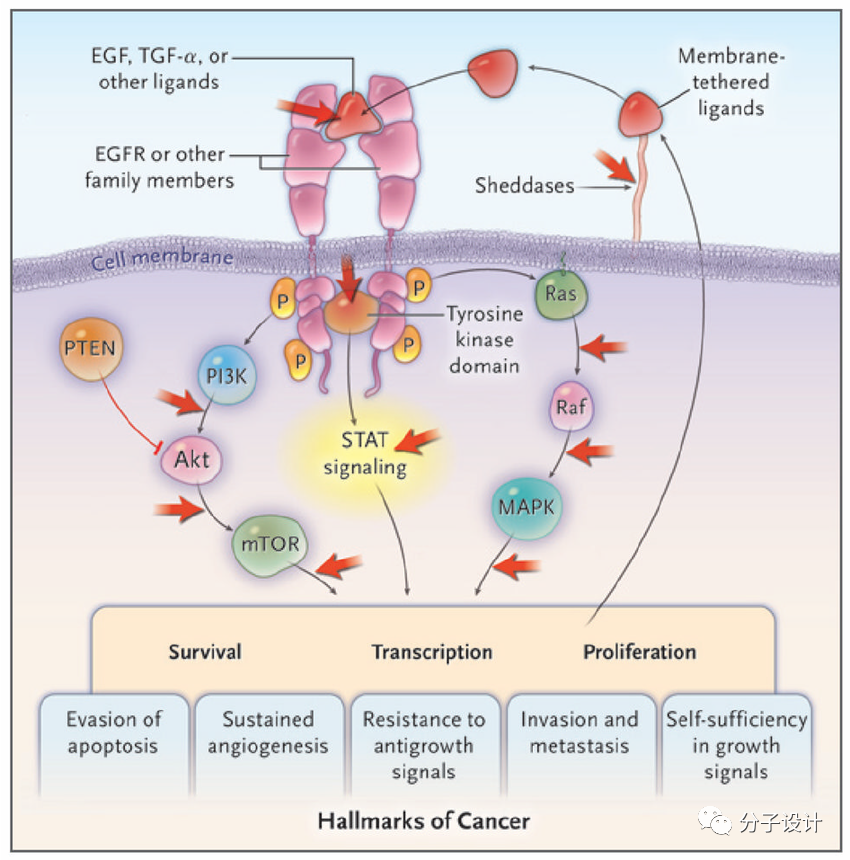
Mutations in the catalytic domain of EGFR will mediate higher kinase activity and ligand independence, such as L858R (single nucleotide substitution in exon 21) and deletion of exon 19 (delE746-A750) in the EGFR coding gene. ), which leads to the beginning and progression of cancer. NSCLC is one of its mutation-induced cancers.
Gefitinib and erlotinib are currently used for the first-line treatment of NSCLC. But after a few months of treatment with these drugs, NSCLC cells developed drug-resistant secondary point mutation T790M. Compared with other carcinogenic mutations, the T790M mutation increases the affinity for ATP, which makes it difficult for existing drugs to bind.
In order to treat patients with drug-resistant NSCLC mediated by the T790M mutation, researchers designed and developed second-generation EGFR-TKIs that covalently bind to the cysteine (Cys797) residues on the EGFR ATP binding site.
Unfortunately, due to targeted dose-dependent toxicity and severe side effects, the efficacy of second-generation EGFR inhibitors at the clinical level is limited. Considering these issues, researchers have developed a third-generation selective and irreversible EGFR inhibitor against mutants.
These inhibitors mostly target the ATP binding pocket of the T790M mutant EGFR and form a covalent bond with Cys797 to overcome the resistance of patients with T790M mutant NSCLC. Although the initial efficacy is impressive, the sensitivity of third-generation EGFR-TKIs is greatly reduced due to the C797S mutation at the edge of the EGFR ATP binding pocket.
At present, the first to third generation inhibitors of EGFR are all designed for the ATP binding site of the EGFR-TK domain. Therefore, it is necessary to find compounds that bind to other sites to overcome the T790M and C797S mutation-mediated effects on EGFR. -TKIs resistance.
In recent years, the fourth generation of EGFR allosteric inhibitors can bind to allosteric sites other than the ATP binding pocket and become a treatment option for mutant NSCLC.
Preliminary studies of these allosteric inhibitors have shown that although they are weak or ineffective when used alone, the combination therapy can exert full efficacy.
1. Overview of EGFR-TKIs
Stanley Cohen of Vanderbilt University won the Nobel Prize for his discovery of epidermal growth factor (EGF) and its receptors. Research on molecules that can inhibit tyrosine kinases began in the 1960s. So far, researchers have designed/discovered four generations of small molecule EGFR-TKIs.
Erlotinib and Gefitinib are the most important two of the first-generation EGFR-TKIs and have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of patients with advanced NSCLC. Erlotinib and gefitinib can reversibly bind to the tyrosine kinase domain of EGFR and inhibit the activation of EGFR and its subsequent downstream signaling pathways.
However, most of the medication patients suffer from the T790M mutation after several months of treatment. It is resistant to the first generation of EGFR-TKIs. The second generation of EGFR-TKIs, such as Afatinib, Dacomitinib and Neratinib, are covalently bound to Cys797 residues in the EGFR ATP binding site to overcome The first-generation EGFR-TKIs T790M mutation-mediated drug resistance.
Due to dose-dependent side effects, such as skin rash and gastrointestinal diseases, the further clinical application of second-generation EGFR-TKIs is limited. Most third-generation EGFR-TKIs are designed to irreversibly target the T790M mutant EGFR, such as osimertinib, rociletinib, and olmutinib, but are affected by the ATP binding pocket C797S is mutated and resistant. recently.
The fourth-generation EGFR-TKIs, such as EAI001 and EAI045, act on allosteric sites far away from the ATP binding site of EGFR, which can overcome the T790M and C797S mutation resistance of the third-generation EGFR-TKIs.
| Generation | Name | Limitations/failure |
| First-generation EGFR-TKIs | Erlotinib Gefitinib Icotinib | Resistance emanation due to T790M mutation, dysregulated EGFR signaling (MET amplification, FGR bypass signaling, HER2 amplification, HGF overexpression, and IGF1R abnormalities) and mutated EGFR downstream signaling (BRAF mutation, K-RAS mutation, loss of PTEN, and PIK3CA mutation). Targeting wild type EGFR leads to severe side-effects dose-dependent toxicity in patients also limited the use of first-generation EGFR-TKIs. |
| Second-generation EGFR-TKIs | Dacomitinib Neratinib Afatinib | During phase II clinical trial patients earlier treated with first generation EGFR-TKIs did not show improvement in their overall survival. Effective and significant results were not observed during phase II clinical trial in EGFR mutated NSCLC patients. During clinical trials no change in overall survival of patients were observed. |
| Third-generation EGFR-TKIs | Nazartinib Naquotinib Olmutinib Rociletinib Osimertinib | Resistance emanation due to EGFR-based mechanisms such as emergence of EGFR mutations (C797S, C797G, E709K, G796, L718, L792, L798, and L692V mutations) and EGFR-independent mechanisms such as dysregulated EGFR signaling (MET amplification, FGR bypass signaling, HER2 amplification, HGF overexpression, and IGF1R abnormalities), and mutated EGFR downstream signaling (BRAF mutation, K-RAS mutation, loss of PTEN, and PIK3CA mutation). |
| Fourth-generation EGFR-TKIs | EAI001 EAI045 DDC4002 DDC-01-163 | Still not approved at clinical level and not significantly effective as an anticancer agent alone in preclinical studies. |
2. EGFR allosteric inhibitor for the treatment of T790M/C797S NSCLC
L858R/T790M and C797S mutations caused the EGFR-TK resistance problem, which promoted the discovery of the fourth generation of EGFR-TKIs. Tyrosine kinase receptors have three binding sites: inactivation site, ATP competition site and allosteric site. Because the ligand or drug cannot bind to the inactivation site, it is impossible to use this as a target; the ATP binding site is currently the binding site of the first to third generation EGFR-TKIs; therefore, the allosteric site is to overcome the mutation Where the hope lies.
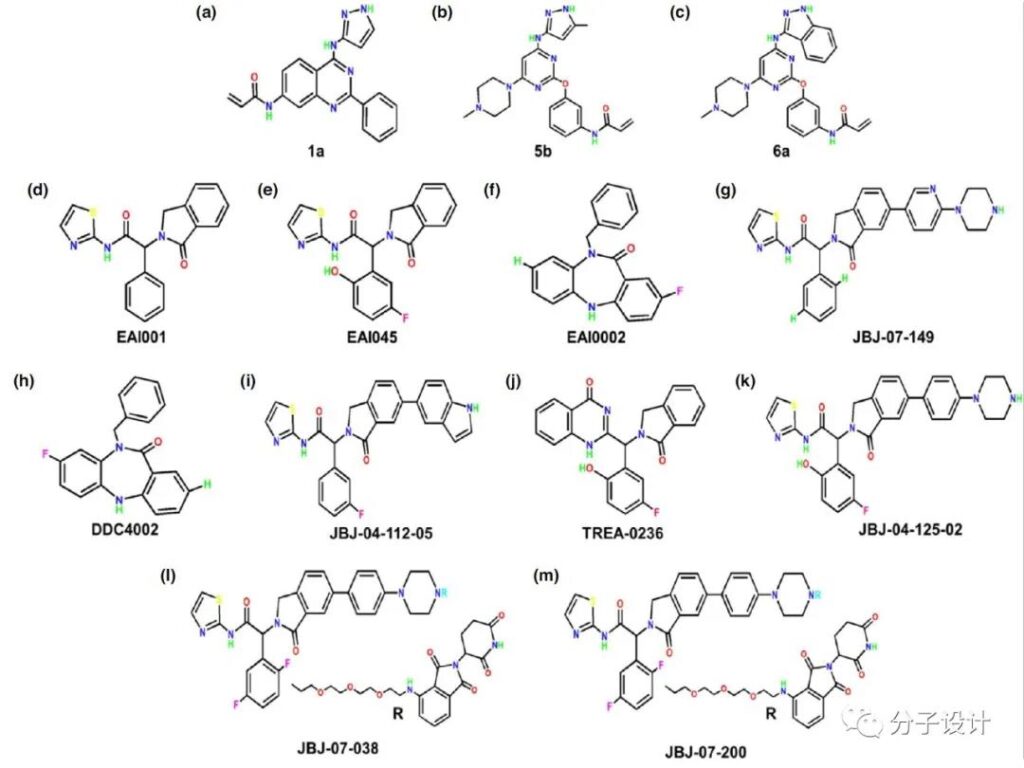
In the initial search for allosteric inhibitors, Engel et al. performed phenotypic screening on 80 NSCLC cell lines and obtained compound 1a that specifically inhibits the drug-resistant H1975 cell line (L858R/T790M). The researchers also designed, screened and optimized a series of new inhibitors, from which 5b and 6a were screened. Jia et al. used the purified target protein L858R/T790M EGFR kinase for biochemical screening and found a non-ATP-competitive fourth-generation EGFR allosteric inhibitor (EAI001).
EAI001 binds to the allosteric site next to the T790M mutant EGFR ATP binding site, and the NH group of its carboxylic acid amide forms a hydrogen bond with Asp855. The IC50 value of EAI001 is 24 nM, which is higher than that of wild-type EGFR (IC50> 50 μM), and the IC50 of EAI001 to L858R and T790M single mutants are 0.75 μM and 1.7 μM, respectively.
EAI001 was further optimized to obtain EAI045, which is about 1000 times more selective for L858R/T790M mutant EGFR (IC50 = 3 nM) than wild-type EGFR. Importantly, EAI045 also has high selectivity for non-kinase targets in its pharmacological safety evaluation. In addition, by attaching the 5-indole substituent to the isoindolinone of EAI001, the researchers also designed a new EGFR allosteric compound JBJ-02-112-05, which has an IC50 for L858R/T790M mutant EGFR Is 15 nM. JBJ-02-112-05 was further optimized to obtain the EGFR allosteric inhibitor JBJ-04-125-02, which has an IC50 of 0.26 nM for T790M/L858R mutant EGFR.
Similarly, for the basic structure of EAI001, Dries developed EAI002, which showed that mutant EGFR has good selectivity compared to wild-type EGFR. With the development of PROTAC technology, a new EGFR mutant (L858R/T790M), a selective allosteric degrader JBJ-07-149, obtained by chemical modification of EAI001, has an in vitro IC50 = 1.1 nM.
On this basis, DDC-01-163, JBJ-07-038 and JBJ-07-200 were further optimized. The results of in vitro biochemical screening showed that, compared with the parent JBJ-07-149, DDC-01-163 has acceptable biochemical activity against the EGFR L858R/T790M mutant (IC50 = 45 nM).
3. The pharmacokinetics of EGFR allosteric inhibitors
Preclinical studies have shown that the fourth-generation EGFR allosteric inhibitor EAI045 has satisfactory stability, bioavailability, excretion and tissue distribution. Jia’s pharmacokinetic analysis of EAI045 shows that in a mouse model, the maximum blood concentration of the drug when taken orally at 20 mg/kg is 0.57 μM, the half-life is 2.15 h, and the oral bioavailability is 26%.
The plasma analysis of oral (30 mg/kg) and intravenous (2 mg/kg) EAI045-treated mice showed that the average 24-hour plasma concentration (Cmax/C5 min) was approximately 1275.802 ng/mL and 1831.146 ng/mL, respectively. The important thing is that EAI045 is rapidly absorbed in various tissues and distributed widely, and the liver is the main organ of its metabolism. In contrast, the recently developed EGFR allosteric inhibitor JBJ-04-125-02 has stronger anti-proliferative activity than EAI045.
JBJ-04-125-02 intravenous injection of 3mg/kg, the half-life of 3 hours is medium, the high area under the curve is 728577 min-ng/mL, but the maximum oral plasma concentration of the compound (20 mg/kg body weight) is 1.1 mmol/L, and the bioavailability is only 3%, suggesting that the compound may begin to accumulate in the patient’s plasma.
If EGFR allosteric inhibitors are to be used as anti-lung cancer drugs, more pharmacokinetic studies are needed.
4. Preclinical and clinical status of EGFR allosteric inhibitors
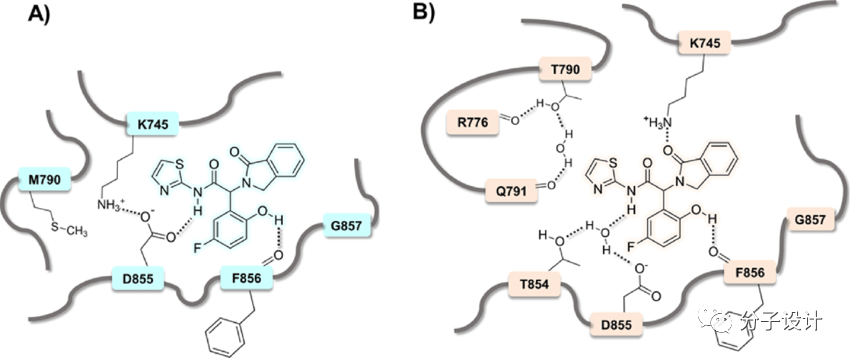
EGFR allosteric compounds EAI001 and EAI045 can overcome the enhancement of ATP affinity mediated by the T790M mutation, and may reduce the autophosphorylation of the T790M mutant EGFR. These compounds bind to the allosteric site of the kinase and provide the stability of the tyrosine kinase domain by turning the regulatory C-helix outward. In preliminary preclinical studies, EAI045 showed good targeting selectivity for L858R/T790M/C797S mutant EGFR.
EAI045 may reduce but not completely inhibit the autophosphorylation of EGFR in L858R/T790M mutant NSCLC cell line H1975 and fibroblast cell line NIH-3T3. The same study confirmed that EAI045 treatment inhibited the phosphorylation of EGFR at Y1173 of the H1975 cell line (IC50 = 2 nM), while the H3255 cell line with the l858R mutation showed moderate activity. EAI045 did not induce the inhibition of wild-type EGFR phosphorylation in HaCaT keratinocytes.
EAI045 has anti-proliferation effect on L858R and L858R/T790M mutant Ba/F3 cells, but has no effect on T790M/exon19del or parental Ba/F3 cells. These results confirm that EAI045 is more selective for mutant EGFR than wild-type EGFR. However, the half of the antiproliferative effect of EAI045 on H1975 and H3255 cell lines requires a concentration of approximately 10 μM, which is much higher than the in vitro IC50 dose.
In terms of in vivo efficacy, EAI045 did not show significant tumor regression in L858R/T790M mutant and L858R/T790M/C797S mutant lung tumor xenograft mice. These findings indicate that EAI045 has limited clinical efficacy as a single drug in the treatment of patients with EGFR-mutant NSCLC.
Another EGFR allosteric inhibitor JBJ-04-125-02 showed strong anti-proliferation and downstream AKT and ERK 1/2 in Ba/F3 cell lines carrying L858R, L858R/T790M or L858R/T790M/C797S mutations Phosphorylation inhibits activity. However, the compound did not show any growth inhibitory activity in parent Ba/F3 or wild-type EGFR Ba/F3 cells, indicating that mutant EGFR has targeting selectivity to wild-type EGFR. Compared with osimertinib, nanomolar JBJ-04-125-02 showed stronger anti-proliferation and tumor suppressive activities in L858R/T790M mutant H1975 cells and tumor transplants, respectively.
The EGFR allosteric degradation agent DDC-01-163 has selective anti-proliferative activity against L858R/T790M mutant Ba/F3 cells (IC50 = 0.096 μM) and wild-type Ba/F3 cells (IC50 >10 μM). Although DDC-01-163 (IC50 = 0.096 μM) is 45 times less potent in biochemical analysis, cell analysis found that DDC-01-163 (IC50 = 0.096 μM) is more potent than its parental allosteric EGFR inhibitor JBJ -07-149 (IC50> 4.9 μM) is 51 times stronger.
Third-level mutations, such as L858R/T790M/C797S and L858R/T790M/L718Q EGFR, are known to develop ossitinib-acquired resistance in non-small cell lung cancer. Interestingly, DDC-01-163 has anti-proliferative activity against Ba/F3 cell lines carrying L858R/T790M/C797S (IC50 = 0.041 μM) and L858R/T790M/L718Q (IC50 = 0.028 μM) EGFR mutations.
The same study confirmed that the degradation rate of DDC-01-163 (0.1 μM) on L858R/T790M/C797S and L858R/T790M/L718Q EGFR mutant cells were 74% and 71%, respectively.
5. Combination therapy strategy of EGFR allosteric inhibitor
In vitro and in vivo studies have shown that the use of EGFR allosteric inhibitors as a single therapeutic agent for the treatment of non-small cell lung cancer may not be so effective. In this case, the allosteric EGFR inhibitor as a combination therapy provides an effective treatment option for NSCLC. In L858R/T790M mutant Ba/F3 cells, although the combination of EAI045 and cetuximab (10 mg/mL) is ineffective, it has a synergistic blocking effect on EGFR dimerization and has an anti-proliferative effect (IC50 = 10 nM). ).
At the same time, oral administration of EAI045 (60 mg/kg/day) and cetuximab (intraperitoneal; 1 mg every other day) induces xenograft tumor models in mice with L858R/T790M or L858R/T790M/C797S EGFR mutant Tumor growth decreased significantly.
EAI045 almost meets all the criteria required for an ideal combination chemotherapy regimen, such as the selectivity of EGFR mutants to wild-type, and the synergy when used in combination with cetuximab.
Another EGFR allosteric inhibitor JBJ-04-125-02 combined with Ositinib is being used to treat EGFR-mediated mutational resistance in non-small cell lung cancer. In osimertinib (0.1 μM) drug-resistant lung cancer cell line (H3255GR), the combined use of 1-10 μM JBJ-04-125-02 significantly increases the pro-apoptotic ability and curative effect of osimertinib. In addition, the combined treatment of JBJ-04-125-02 (100 mg/kg) and osimertinib (2.5 mg/kg) has a much greater inhibitory effect on tumor growth in xenograft mouse models than the two drugs alone.
Similarly, the recently developed allosteric EGFR degradant DDC-01-163 (0.01 μM and 0.1 μM) synergistically enhances the anti-proliferation and apoptosis ability of osimertinib (0.01 μM) at a certain dose. When allosteric inhibitors are used in combination with traditional drugs, a significant synergistic effect can be observed, that is, the therapeutic potential of allosteric drugs. This indicates that allosteric EGFR inhibitors may enhance the clinical efficacy of traditional EGFR-TKIs in the treatment of EGFR-mutant NSCLC.
This requires more preclinical and clinical studies to verify the effect of allosteric EGFR inhibitors as a combination therapy for non-small cell lung cancer.
Future prospects of EGFR allosteric inhibitors
Although the therapeutic effect of EGFR-TKIs (first generation to third generation) has been substantially improved, they still cannot bring sufficient therapeutic relief to NSCLC. EGFR mutations (T790M and/or C797S) lead to the loss of the ability of EGFR-TKI to bind to the EGF receptor, leading to inevitable resistance.
The results of the study show that the allosteric site of epidermal growth factor receptor has the potential to inhibit the proliferation of cancer cells. At the same time, targeting EGFR allosteric sites provides an efficient and selective new treatment strategy for the treatment of EGFR mutant NSCLC.
The T790M and C797S mutations in the active site of EGFR do not affect the efficacy of EGFR allosteric inhibitors because they target the allosteric site of EGFR, which occurs outside the ATP binding site.
However, these inhibitors are currently in the development stage, and due to receptor dimerization, they are not particularly effective as a single drug for EGFR mutant NSCLC.
In this case, the combined use of EGFR allosteric inhibitors and conventional chemotherapy or immunotherapy is an efficient strategy to overcome EGFR mutation-induced NSCLC resistance.
Existing studies have shown that the combined treatment of EGFR allosteric inhibitors and chemotherapy or immunotherapy is more effective than using these two drugs alone.
Finally, the prospect of combined therapy of EGFR allosteric inhibitors is rapidly emerging; however, more preclinical and clinical studies are needed to further confirm their combined treatment of EGFR mutant-resistant NSCLC patients.
(source:internet, reference only)
Disclaimer of medicaltrend.org



