How to optimize the CAR-T cell manufacturing process during clinical trials
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
How to optimize the CAR-T cell manufacturing process during clinical trials?
How to optimize the CAR-T cell manufacturing process during clinical trials ? Tisagenlecleucel is a CD19-specific chimeric antigen receptor (CAR)-T cell therapy, approved for relapsed or refractory B-cell precursor acute lymphoblastic leukemia (B-ALL) and Relapsed or refractory diffuse large B-cell lymphoma (DLBCL).
The initial tisagenlecleucel manufacturing process technology was developed in an academic center, and then transferred, optimized, validated, and expanded to provide large-scale global trials before commercialization.
Tisagenlecleucel produced in two centralized facilities has been successfully used in global multi-center trials of B-ALL and DLBCL (more than 50 clinical centers in 12 countries).
In this article, we describe some continuous process improvements made to Tisagenlecleucel manufacturing while maintaining and improving product quality in order to meet stable demand over time.
These enhancements improve manufacturing capacity, process stability, manufacturing success rate and product quality, and shorten production time.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy involves reprogramming the patient’s T cells to target and attack tumor cells1,2, and it has been successfully used in clinical trials for the treatment of B cell malignancies [3-7] .
Tisagenlecleucel (Kymriah; Novartis, Basel, Switzerland) is the first CD19-specific autologous CAR-T cell product approved by the U.S. Food and Drug Administration (FDA) and the European Commission for the treatment of R2 lines of type B patients under 25 years of age After systemic therapy, acute lymphocytic leukemia (B-ALL), the cell precursor of refractory or secondary or secondary relapse and adult patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL) [8, 9].
Another CD19 CAR-T cell product, axicabtagene ciloleucel (Yescarta; Kite Pharma, Santa Monica, CA, USA) has also been approved by the FDA and the European Commission. After the R2 line of systemic therapy, it can be used to treat patients with relapsed or difficult Adult patients with therapeutic DLBCL [10,11]. Several other CAR-T cell products are also in various stages of clinical development [12].
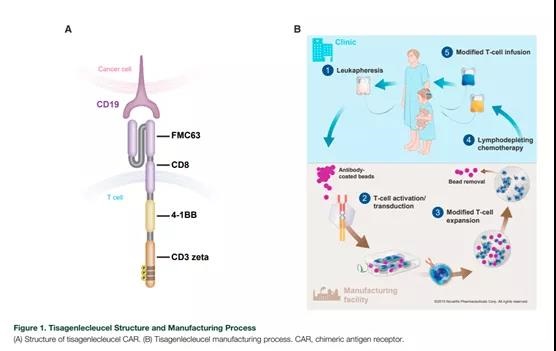
The tisagenlecleucel CAR contains a murine single-chain antibody fragment specific for CD19 (FMC63), followed by the CD8-a hinge and transmembrane region, fused with the intracellular CD3-z signal domain and 4-1BB costimulatory domain (Figure 1A).
The mechanism of action of Tisagenlecleucel has been described previously [3,13]. The 4-1BB domain is a costimulatory signal for T cell activation, which is important for the expansion, persistence and anti-tumor activity of Tisagenlecleucel [3,13,14].
Tisagenlecleucel experienced expansion after infusion of patients. When CAR binds to CD19 on target cells in vivo, it initiates signal transduction, leading to T cell activation, cytokine production and target cell destruction.
In clinical trials, the strong expansion and long-term persistence of tisagenlecleucel, as well as the long-lasting clinical response after infusion were confirmed in patients with B-ALL, chronic lymphocytic leukemia and DLBCL [5,15-17].
The initial manufacturing process of tisagenlecleucel was developed at an academic center at the University of Pennsylvania (Philadelphia, Pennsylvania, USA). The process was then transferred to Novartis, which was then optimized and expanded to provide global clinical trials. In order to meet the growing demand, resolve differences between donors and improve product quality, processes must be optimized and improved.
These improvements focus on enhancing compliance, assisting scalability, and allowing for global distribution. These improvements have led to the current manufacturing process.
Tisagenlecleucel is produced in two centralized chemical plants and has been successfully used in global clinical trials (ELIANA [ClinicalTrials.gov: NCT02435849] 5 and JULIET [ClinicalTrials.gov: NCT02445248] 6), involving more than 50 clinical centers in 12 countries. In this article, we describe the tisagenlecleucel manufacturing process, the initial challenges encountered in the horizontal expansion process and the continuous improvement in key trials to finally solve the challenges of the tisagenlecleucel commercial production process.
Tisagenlecleucel manufacturing process
The production process of tisagenlecleucel (Figure 1B) begins with the collection of unmobilized peripheral blood mononuclear cells from the patient through leukocyte separation.
After collecting the white blood cells, the material should be frozen and stored within 24 hours after collection, and stored below -120°C. The cryopreserved material is then transported to the manufacturing plant and stored below –120°C until it is ready for further processing.
When the manufacturing tank is available, the patient’s leukocyte separation technique material is thawed under controlled conditions, and then the cells are washed to remove the freezing liquid.
Use anti-CD3/CD28 antibody-coated paramagnetic beads to enrich, select and activate T cells, and then transduce them with a self-inactivating lentiviral vector containing an anti-CD19 CAR transgene [2,13].
After transduction, the excess vector and other residues are washed away from the culture, and the cells are expanded in a static culture and then in a bioreactor.
Cell expansion continues in vitro until there are enough cells to meet the dosage requirements of the final product. For cell harvest, the transduced T cells are separated by separating the transduced T cells from the beads, washed, prepared in an insoluble medium, transferred to an infusion bag and frozen for storage.
The critical quality attribute (CQA) test of tisagenlecleucel was carried out (Table 1). After the final product was released, the cryopreserved tisagenlecleucel was transported to the treatment site.

The development history of tisagenlecleucel manufacturing process
The early studies of tisagenlecleucel enrolled relatively few patients, and the products used in these studies were produced at the University of Pennsylvania using the previously described process [3,4,18-20]. Transition from a flexible process of a single academic institution to a global study.
This study recruited patients from many clinical sites and manufactured them in two centralized manufacturing plants that eventually led to commercial manufacturing, which required many blood separation and treatment sites as well as manufacturing Extensive coordination between factories [2].
The initial process includes (1) the standardization of the overall manufacturing process and the description of the characteristics of the process and products; (2) meeting the specific regulatory requirements of various countries and regions at the same time; (3) changing from a manual process to a verifiable automated manufacturing solution; ( 4) Manage complex logistics during the transition from phase 1/2 clinical trials to critical global trials. Through the collaboration of various technology transfer teams from academia, manufacturing, technology development, quality assurance, and regulatory agencies, a method of gradually implementing the technology transfer process was developed (Figure 2).
This cooperation successfully transferred the production process of tisagenlecleucel from academia to industry.
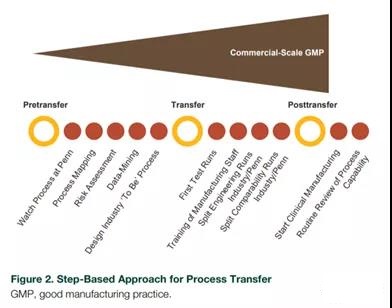
Transfer of Tisagenlecleucel production
During the transition from academic manufacturing to industrial manufacturing for global clinical trials, steps have been taken to improve process performance and durability while maintaining the quality of Tisagenlecleucel products (Table 1).
Some key process changes include enhancing process control to ensure consistency, replacing manual processes with automation to ensure repeatability, validating analytical methods to improve consistency, and using closed systems to prevent contamination.
The recognition of method validation and implementation of more robust and/or faster methods to improve the performance of analytical methods is a key area during technology transfer and horizontal promotion. A new quantitative method for CAR protein expression using anti-idiotypic antibodies improves the robustness and linear range of the measurement method used for dosimetry. The optimization of the vector used in the CAR-T cell transduction step helps reduce variability, maximize the transduction efficiency, and allow the scale of the manufacturing process to be expanded. The production process of lentiviral vectors has also been improved, and the vectors can be produced on a large scale under Good Manufacturing Practice (GMP) conditions [2].
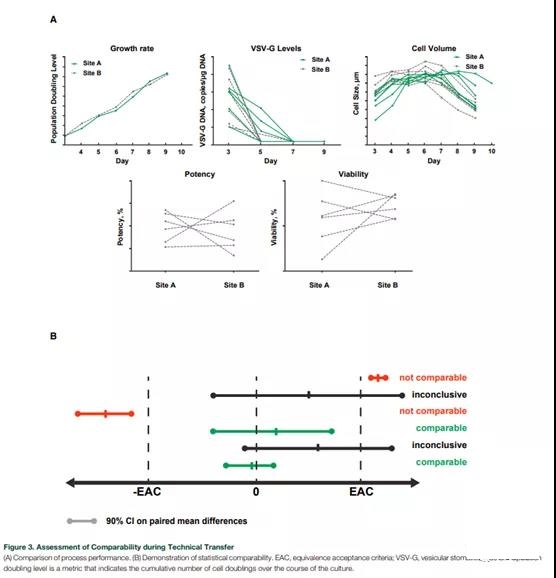
The comparability of process performance and product quality during technology transfer is established by evaluating selected parameters (such as cell growth rate, cell size, potency, viability, and removal of carrier residues) (Figure 3A). Equivalence tests were conducted for each CQA to prove the comparability of the analysis (Figure 3B). The setting of the equivalent acceptance standard depends on the understanding of the manufacturing process, and may be based on the variability of the method and/or process.
Compared with fresh materials, cryopreserved leukocyte separation is selected as the preferred raw material to meet the needs of centralized production in global trials, while maximizing the flexibility of patient management.
Cryopreservation of leukocyte separation can extend the storage time of raw materials in the case of manufacturing delays, achieve reliable and consistent production, and most importantly, provide patients with the flexibility of leukocyte separation at the most convenient and patient-friendly time .
Rather than being consistent with the availability of manufacturing slots. Extensive evaluation of the cryopreservation process (including cryopreservation media, freezer bags, storage, freezer transport, thawing conditions and the stability of leukocyte separation materials after snow melting), and optimized and verified the manufacturing process using cryopreserved starting materials, It can be used for pediatric and young adult patients with B-ALL and adult DLBCL patients.
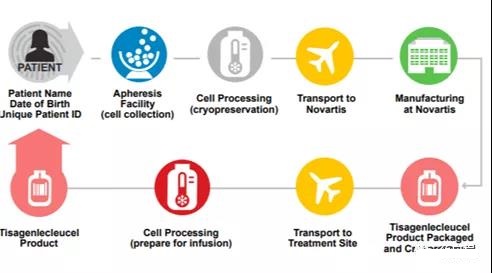
Logistic controls are planned and implemented to meet expected patient needs. Both suppliers and service providers comply with GMP standards and have established partnerships to ensure the long-term supply of key raw materials. A system to control the chain of custody was developed using custom software to track the patient’s identity during the return of leukocyte separation materials from the clinic to the production Tisagenlecleucel, and to ensure that the patient is injected with its own cells (Figure 4).
Formal process verification was carried out in clinical trials. The CQA of tisagenlecleucel was identified based on the standard test of patient safety and the specific markers of clinical efficacy of the product. A risk-based approach was used to characterize process performance and identify key control parameters. A reliable product release process was established to verify the safety, purity, homogeneity and efficacy of each patient-specific Tisagenlecleucel product (Table 1) according to commercial standards.
Early challenges in the clinical trial of Tisagenlecleucel and improvement of subsequent processes
In the early days, logistical challenges related to meeting the manufacturing needs of the multicenter trial and adapting to the observed differences in leukocyte separation materials among patients led to higher than expected failure rates between enrollment and infusion. In order to solve these challenges, as described below, enhancements have been made to improve manufacturing capacity, process durability, process success rate and product quality, and reduce production time.
The combination of manufacturing experience gained in clinical trials and process characteristics can help identify specific areas for improvement. For example, the initial process of T cell enrichment uses multiple pathways to adapt to changes in the composition of the cell population of incoming patient leukocyte separation materials.
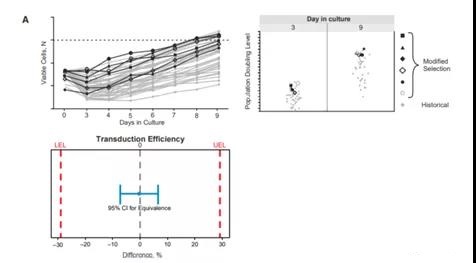
Experimentation and evaluation of different options led to the optimization of the T cell enrichment step into a single improved pathway that can be used for a series of incoming cell populations. These modifications to the T cell enrichment process lead to streamlined manufacturing with improved cell growth kinetics and equivalent product quality.
As shown in Figure 5A, the cell growth curve and population doubling are comparable using this improved selection method compared with the historical method. Based on equivalence tests, the comparability of these methods has been proved.
Equivalence tests have shown that the transduction efficiency of cells generated using the improved process is equal to or higher than that of cells generated using the historical process.
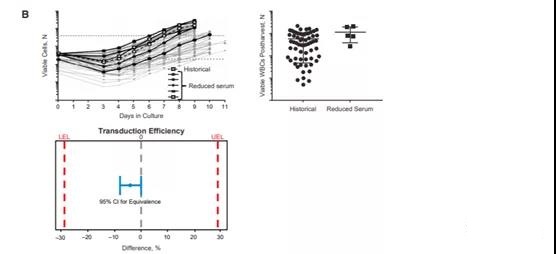
The amount of human AB serum used in the culture medium has been identified as a potential rate-limiting step and needs to be reduced to overcome global supply constraints. Reducing the serum concentration in the culture medium can achieve a more sustainable process with the same growth kinetics and product quality. As shown in Figure 5B, the cell growth profile and the number of viable white blood cells in the reduced serum batch are within the range of the historical manufacturing run. Equivalence tests have proved that the transduction efficiency of cells generated using different processes is comparable.
Another rate-limiting step in the manufacturing process is the time required for sterility testing by standard pharmacopoeial methods. The verification and use of rapid quantitative Mycoplasma sterility analysis based on polymerase chain reaction reduces the time required for sterility testing before product release. Strict sterility testing during the production of Tisagenlecleucel is part of the overall sterility assurance strategy of the aseptic system to ensure that Tisagenlecleucel does not contain potential contaminants. This proven method ensures that quality standards meet regional and global regulatory requirements.
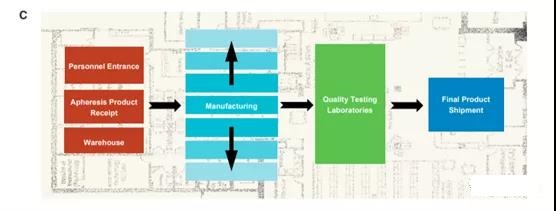
In order to meet global clinical needs, facilities and operations must be improved. These improvements include measures to increase production capacity, reduce production time and reduce patient dropout rates. The staffing has been optimized to receive the increased injection volume, while the clean room capacity increases accordingly.
The modular facility design can be flexibly expanded horizontally along with the continuously evolving manufacturing platform form, and has the ability to manufacture a variety of indicating products (Figure 5C).
As the needs of patients increase, processing time is reduced by optimizing training plans, increasing staff, shifts, and optimizing processes and logistics. An automatic electronic ordering system was introduced to ensure the collection of white blood cells, to maintain the identity chain and the chain of custody throughout the manufacturing process, and finally to transport the manufactured products to the clinical site.
The transportation logistics is evaluated, which is essential to ensure that patients receive medication in a timely manner. The evaluation also includes the design of refrigerated transport machines, temperature control mechanisms and couriers who provide transportation services.
Identifying areas for improvement and addressing these issues through process improvements supported by appropriate research will help optimize the manufacturing process during critical trials.
Recently, variability in the percentage of viable cells in commercial products produced for adult DLBCL patients has been observed due to the increased variability of incoming patient materials. Despite this variability, most of these products can still be administered to patients.
Successful manufacturing, short production time
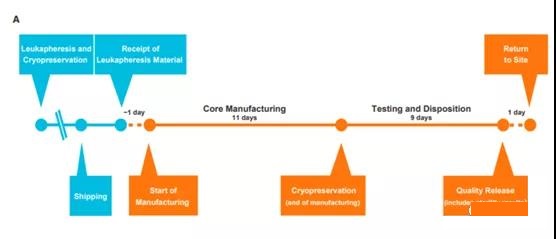
Optimized commercial production of Tisagenlecleucel In ongoing clinical trials, continuous improvements in the manufacturing process have led to a consistent and optimized process for Tisagenlecleucel that can adapt to the changes observed in incoming leukocyte separation materials during the trial (Figure 6A ).
In this way, a 22-day goal can be set for the manufacturing cycle, and obtain a very high manufacturing success rate in a commercial environment. These changes are first evaluated in development studies and then introduced in clinical trials after the comparability of the changed products is proven.
This is essential to ensure that patients get products of consistent quality. According to the first batch of 37 commercially produced Tisagenlecleucel products for B-ALL (deadline, January 30, 2018), the current process shows that the median throughput time from receiving leukocyte removal to 2007 is 23 days ( The range is 21-37 days). Manufacturing plant to return Tisagenlecleucel to the clinical site (including transportation time; Figure 6B).
During this period, commercial patient orders came from 13 treatment locations in a large area of the United States (Figure 6C). It is expected that ongoing process improvements will further reduce manufacturing production time.
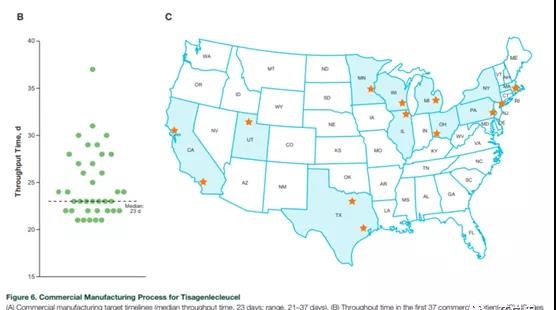
Patients younger than 3 years old were excluded from the ELIANA study; 5 However, the youngest patient to produce tessalyl ribonucleic acid in a commercial environment was 2 years old. The patient’s preparation and reinfusion time was 24 days. The production of patients under 3 years of age will continue to be monitored and evaluated to ensure that T cells are available to start production and to ensure the ability to provide appropriate doses to such young patients.
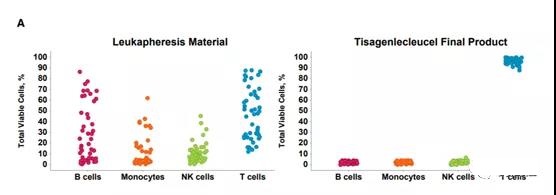
Tisagenlecleucel product characteristics
Extensive product testing proves the consistency of product quality during the manufacturing process. The evaluation of the T cell population in the ELIANA trial (Figure 7A) showed that the T cell product developed from the variable patient leukocyte separation starting material was consistent.
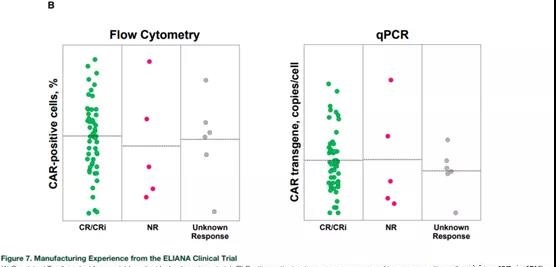
The analysis of CAR expression and transgene copy of each cell showed that the vector integration was stable, and positive clinical results were observed within the allowable range (Figure 7B).
Positive clinical results have also been observed across the entire range of acceptable potency determination results, and the clinical response, safety and in vivo expansion during the clinical study are not affected by the characteristics of the selected product [16,21].
Conclusion and future direction
Tisagenlecleucel’s current manufacturing process reflects how to successfully expand, streamline and optimize cell therapy with complex manufacturing processes to ensure the delivery of high-quality products to the global patient population.
By focusing on key areas of improvement, without affecting product integrity or performance, the production process of Tisagenlecleucel has been optimized and production efficiency has been achieved. In the global multi-center trials, Tisagenlecleucel has accumulated considerable experience in centralized manufacturing.
Through continuous evaluation and improvement, a stable and stable Tisagenlecleucel commercial production process has been developed. The current production process is based on the first 37 commercial patients (for B-ALL), resulting in an average time from receipt of leukocyte removal material from the manufacturing plant to the return of manufactured Tisagenlecleucel to the clinical site of 23 days, including transportation time.
It is foreseeable that ongoing, continuous process improvements will further increase the increase in the production of Tisagenlecleucel and further shorten the production time from receiving leukocyte separation materials to returning finished products.
(source:internet, reference only)
Disclaimer of medicaltrend.org



