How to clear residual cancer cells resistant to targeted therapy?
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
How to clear residual cancer cells resistant to targeted therapy?
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
How to clear residual cancer cells resistant to targeted therapy?
“Science Translational Medicine”: A method to clear residual cancer cells resistant to targeted therapy has been found.
Different from chemotherapy that “kills a thousand enemies and destroys eight hundred”, oncogene-targeted therapy is called a “biological missile” because of its precise effect on cancer-causing targets.
In recent years, a variety of targeted therapy drugs have been successfully used in clinical practice, however, the actual benefits of patients with advanced tumors are not ideal, and one of the important reasons is drug resistance. However, little is known about the mechanisms of acquired resistance to targeted therapy.
Some studies have found that residual cancer cells that survive targeted therapy and subsequent DNA damage repair are important reasons for tumor cells to develop drug resistance [1]. Therefore, exploring DNA damage repair-related molecules induced by targeted therapy and clarifying the mechanism of tumor drug resistance play a crucial role in the development of targeted drugs and the selection of clinical programs.
Recently, Dr. Kris C. Wood of Duke University and his team found that there are DNA double-strand breaks (DSB) and DNA double-strand break repair in tumor cells that survived targeted therapy. Ataxic telangiectasia mutant (ATM) enzyme .
They also found that targeted therapy in combination with an ATM inhibitor eradicated surviving residual tumor cells in cell lines in vitro and in animal models, resulting in a more durable therapeutic response [2].
This research result has laid a theoretical foundation for the integration of clinical development of ATM inhibitors and existing targeted therapy programs . The related research results were recently published in the famous journal Science Translational Medicine .

Next, let’s see how this research was carried out.
First, to investigate whether targeted therapy activates DNA damage response signaling (DDR), the research team used a panel of tumor cell lines sensitive to cognate targeted therapy : epidermal growth factor receptor (EGFR) mutant non-small cells Lung cancer (NSCLC) cell line (PC9, HCC827), anaplastic lymphoma kinase (ALK) gene rearranged NSCLC cell line (H3122), BRAF gene mutant melanoma cell line (A375), FLT3 gene mutant acute myeloid cell line Leukemia (AML) cell line (MOLM13), KRAS gene mutant pancreatic cancer cell line (MIA PaCa-2) and NSCLC cell line (A549).
They found that the expression of phosphorylated ATM (p-ATM) and γH2AX at the S1981 site was significantly increased after treating the above cell lines with different doses of cognate targeted therapeutics for 24 hours .
It should be pointed out that ATM S1981 phosphorylation is a necessary site for activation of downstream DNA damage response signaling pathways, and γH2AX produced by phosphorylation of H2AX (histone H2A family member X) is a biomarker of DNA double-strand breaks .
Subsequent cell clone formation experiments showed that a significant increase in γH2AX expression was still observed at drug doses that did not affect cell viability, a conclusion that was observed in three gefitinib-resistant EGFR-mutant NSCLC cell lines (PC9R , GR4, WZR12) were further validated; meanwhile, negative Annexin V staining (for detection of apoptosis) was observed in PC9 and A549 cells treated with targeted drugs.
Finally, the neutral comet assay (a technique that detects DNA damage at the single-cell level) found that the presence of DNA double-strand breaks was detected in PC9 and A549 cells after 24 hours of targeted drug treatment, suggesting that the observed activation of ATM is due to DNA damage.
These results suggest that tumor cells that survive targeted therapy develop DNA double-strand breaks and subsequent ATM activation .
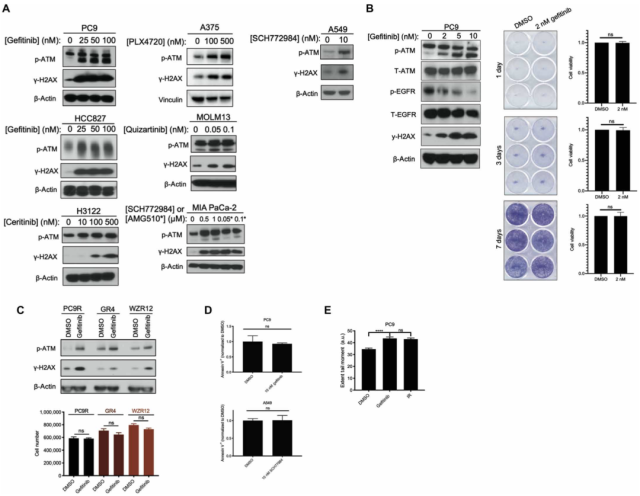
DNA damage response signaling pathway is activated in targeted drug-treated tumor cells
So, how is ATM activated? Let’s look down.
First, PC9 and HCC827 cells were treated with gefitinib (EGFR blocker) for 24 hours, and an increase in the phosphorylation expression of ATM and its substrate checkpoint kinase 2 (Chk2) at T68 was observed; subsequently, the researchers used Treating PC9 cells with an ATM kinase inhibitor (AZD0156) alone or in combination with gefitinib, AZD0156 and gefitinib were found to abrogate γH2AX expression and activation of the DNA double-strand break repair pathway .
In the following experiments, the researchers found that with the increase of gefitinib dose and treatment time, the expression of p-ATM and γH2AX also gradually increased, while the initiator caspase 9 (initiator caspase 9) and Both executor caspase 3 were cleaved.
A recent study showed that activation of endogenous pathway caspases can lead to the formation of DNA double-strand breaks and subsequent activation of ATM [3].
We therefore hypothesized that Bcl-2 anti-apoptotic family members BIM and BAK/BAX activate downstream caspases, and the resulting increase in mitochondrial outer membrane permeability (MOMP), possibly due to DNA double-strand breaks and Reason for ATM activation.
To test this hypothesis, they knocked down the expression of BIM and BAX, respectively, in PC9 cells, and found that low expression of BIM and BAX abolished targeted therapy-induced activation of ATM and γH2AX .
To further test this hypothesis, the researchers treated PC9 cells with a pan-caspase inhibitor (Q-VD-OPh) alone or in combination with gefitinib and found that the combination of the two eliminated gefitinib alone ATM activation observed upon Ni treatment.
Simultaneous knockdown of caspase 3 and 7 in PC9 cells also abolished the activation of ATM and γH2AX in gefitinib-treated PC9 cells. Caspase-activated deoxyribonuclease (CAD), a key enzyme activated by caspase 3 and 7 to mediate DNA double-strand breaks, is expressed in PC9 cells treated with gefitinib How?
The results showed that the expression of deoxyribonuclease was relatively unchanged, but the expression of inhibitor of caspase-activated deoxyribonuclease (ICAD) was significantly reduced; the researchers further knocked down the expression of deoxyribonuclease in PC9 cells and found that gemfi Activation of γH2AX was not observed in tinib-treated PC9 cells , but cleavage of caspase 3 and loss of DNase inhibitors were still observed.
The above results indicate that low expression of DNase inhibitor leads to activation of DNase, which in turn induces DNA double-strand breaks and activation of ATM and activation of caspase and loss of DNase inhibitor in deoxyribonuclease Enzyme activation works upstream .
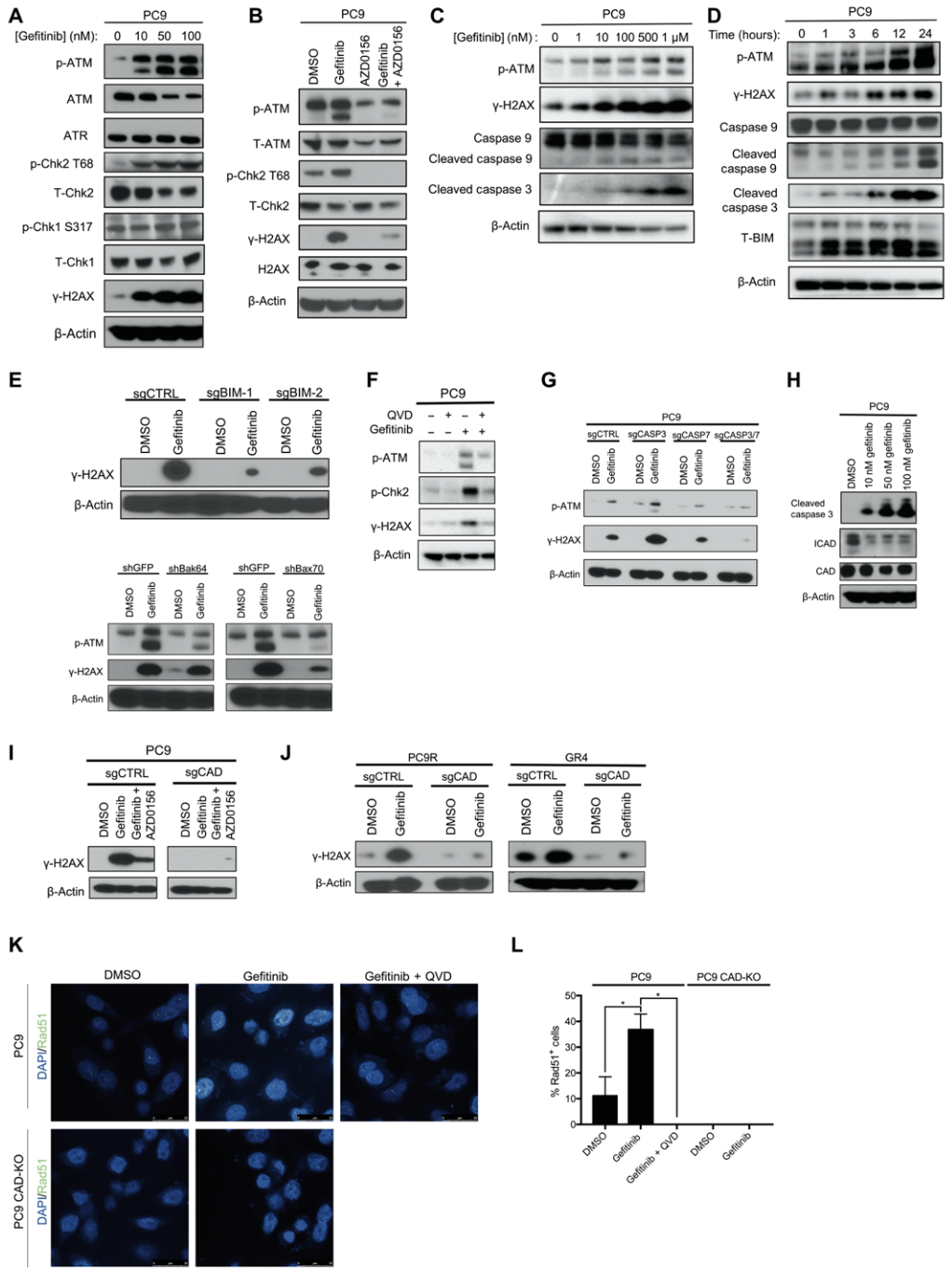
EGFR inhibitor induces activation of ATM signaling pathway
The above results suggest that cells that survive targeted therapy may require ATM activation to repair DNA double-strand breaks caused by targeted therapy exposure. So could ATM inhibitors improve the intensity and duration of responses to targeted therapy?
First, previous experiments have confirmed that AZD0156 can block gefitinib-induced ATM activation, and then researchers treated PC9, PC9R, and GR4 cells with AZD0156 alone or in combination with gefitinib and found that AZD0156 could simultaneously increase resistance to targeted drugs .
Sensitivity of sensitive cell lines (PC9) and targeted drug-resistant cell lines (PC9R, GR4) to gefitinib .
This indicates that residual tumor cells after EGFR inhibitor treatment are still sensitive to the combined use of EGFR inhibitors and ATM inhibitors, and this combination therapy regimen can eliminate tumor cells that survived EGFR inhibitor monotherapy. Next, the research team further verified this conclusion.
A small effect of AZD0156 on cell growth was observed in long-term qualitative time-to-progression (TTP) analysis, whereas gefitinib monotherapy resulted in increased tumor cell resistance around 40 days, while gefitinib and AZD0156 combined Treatment effectively removes residual tumor cells, thereby avoiding the growth of drug-resistant tumor cells .
Finally, the research team assessed the effect of DNase on cellular responses when combined with EGFR inhibitors and ATM inhibitors. The study found that knockdown of DNase expression in PC9 and GR4 cells reversed the inhibitory effect of combination therapy on tumor cell growth.
Next, after inhibiting the expression of ATM, it was found that depletion of ATM abolished gefitinib-induced activation of γH2AX, suggesting that AZD0156 acts by targeting ATM inhibition.
Taken together, these results demonstrate that DNase-mediated DNA double-strand break formation in tumor cells that survive EGFR inhibitor treatment is synthetically dependent on ATM, a key kinase that resolves this DNA damage .
Therefore, the combined use of EGFR inhibitors and ATM inhibitors can eliminate tumor cells that survive EGFR inhibitor monotherapy, thereby inhibiting the growth of drug-resistant tumor cells.
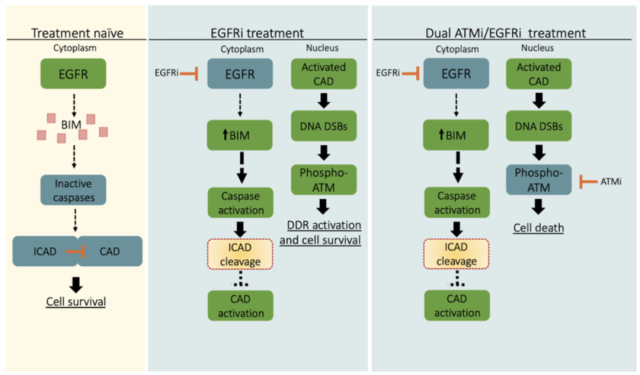
Schematic diagram of the mechanism
In vitro cell experiments have confirmed that the combination of EGFR inhibitors and ATM inhibitors can inhibit the growth of drug-resistant tumor cells. So, will this treatment combination prevent tumor growth in vivo? Next, the research team conducted in vivo xenotransplantation experiments, and let’s see what the results are.
The study found that ATM inhibitors alone had little effect on tumor growth , EGFR inhibitors suppressed tumor growth before tumors developed resistance, and mice treated with a combination of ATM inhibitors and EGFR inhibitors showed sustained growth throughout the study period. tumor regression .
Subsequently, the research team used three cell models extracted from tumor tissues of lung cancer patients, MGH134 (from EGFR-mutant NSCLC patients and resistant to first-line erlotinib treatment) , MGH1109 (from EGFR-mutant NSCLC naïve patients) , MGH006 (from EML4-ALK variant 1 mutant NSCLC treatment-naïve patients) was further validated.
The results showed that ATM inhibitors could suppress the growth of tumors resistant to targeted therapy ; consistent with this finding, in MGH134 xenograft mice, osimertinib treatment produced an initial growth inhibition followed by tumor progression, whereas Combination therapy with osimertinib and AZD0156 produced sustained tumor regression .
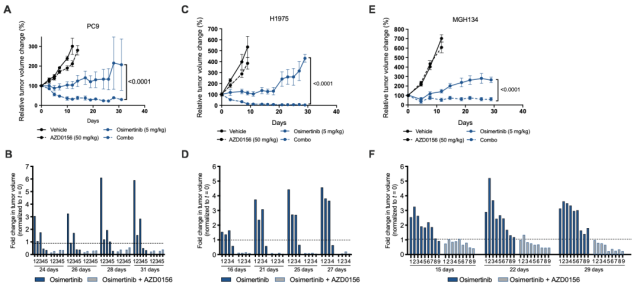
Targeted therapy induces ATM activation in a mouse xenograft model
Finally, in order to explore the possible clinical relevance of these experimental results, the research team analyzed ATM activation in tumor tissue from patients treated with EGFR inhibitors.
First, the researchers performed immunohistochemical (IHC) staining on tumor tissue from five EGFR-mutant NSCLC patients, and the results showed that p-ATM expression was significantly higher in tumor tissue during treatment than before treatment with the EGFR inhibitor erlotinib Increased ; a further comprehensive analysis of tumor samples from all patients found that p-ATM expression was significantly increased in tumor tissue from progressive patients treated with erlotinib.
Next, we analyzed time-to-clinical progression data from the Sloan-Kettering Actionable Cancer Targets (MSK-IMPACT) Clinical Sequencing Cohort database for 11 patients receiving first-line erlotinib who also developed EGFR mutations / Erlotinib sensitizing mutation and ATM mutation .
The results of the analysis showed that EGFR-mutant NSCLC patients with ATM loss-of-function mutations had prolonged progression-free survival compared with EGFR-mutant NSCLC patients without ATM mutations .
The time to disease progression (TTP) was 17.8 ± 10.9 months with erlotinib in patients with tumors with ATM loss -of-function mutations, compared with 9.0 ± 1.9 months in those with tumors that may have non-functional ATM mutations months; this result is consistent with the time to clinical progression of 8 to 12 months observed in multiple studies of unselected patients with EGFR-mutant NSCLC receiving first-line erlotinib [4].
These data above suggest that ATM activation occurs in tumor cells that survive EGFR inhibitor treatment and may play a tumor-protective role.
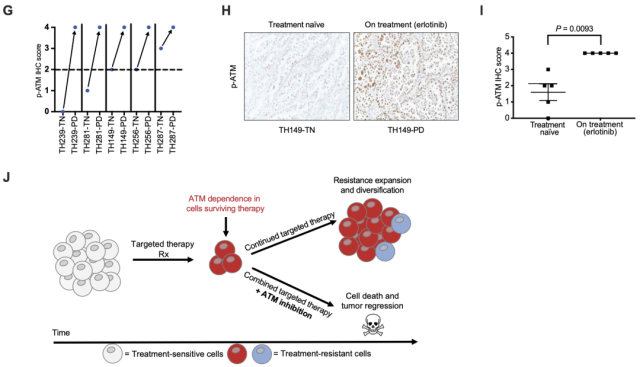
Targeted therapy induces ATM activation in patient tumor tissue
In general, this study explored the molecular mechanism of drug resistance to targeted therapy, and further verified the clearance effect of ATM inhibitor and targeted drug combination therapy on drug-resistant tumors through in vitro and in vivo experiments, and finally returned to the clinic.
The expression of NSCLC patients in tumor tissues before and after targeting, and the effect of ATM loss-of-function mutations on the efficacy of targeted drugs were detected.
This study lays a theoretical foundation for the integration of clinical development of ATM inhibitors with existing targeted therapy programs, as well as the design of clinical trials, and also provides new clinical ideas for diagnosis and treatment.
references
1. doi:10.1126/scitranslmed. aal5148
2. doi:10.1126/ scitranslmed.abc7480
3. doi:10.1038/cr. 2017.41
4. doi: 10.1158/1078-0432.CCR-12-2246
How to clear residual cancer cells resistant to targeted therapy?
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



