New strategies for overcoming drug resistance in targeted tumor therapy
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
New strategies for overcoming drug resistance in targeted tumor therapy
New strategies for overcoming drug resistance in targeted tumor therapy. In the past two decades, drug development in the oncology field has mainly focused on molecularly targeted drugs.
However, with the widespread use of targeted drugs in the clinic, the rapid emergence of drug resistance caused by target mutations has severely reduced the efficacy of the drug. How to overcome drug resistance has become a major problem faced by current anti-tumor drugs.
Major pharmaceutical companies and scientific research institutions are committed to clarifying specific mutations or other genetic/cytological changes that lead to drug resistance, and provide basic support for the development of more effective treatment strategies.
In recent years, drugs including non-classical combination models have emerged , Proteolysis Targeting Chimeras (PROTACs) drugs, targeted allosteric drugs (Allosteric Drugs), multi-site drugs and other strategies to limit the emergence of drug resistance.
In addition, in the early stages of drug development, the combination of drug resistance analysis can also help design and evaluate treatments that have long-term benefits for patients.

From a mechanism point of view, chemical small molecule inhibitors have the function of selectively blocking their targets. They can not only be used as probes to evaluate potential therapeutic strategies based on cell dynamics, but also can be used as the starting point for drug development.
Multiple anti-tumor agents are listed on the market The drugs all come from here. For example, abnormal BCR-ABL fusion signal or up-regulation of epidermal growth factor receptor (EGFR) kinase mutant activity in lung cancer is closely related to the occurrence and development of leukemia. Blocking its activity can effectively improve clinical results.
These studies have brought The upsurge of BAR-ABL/EGFR targeted drug research and development. These targeted drugs have played a significant role in the treatment of leukemia after being marketed. However, soon, most patients will inevitably develop drug resistance to these drugs.
The emergence of drug resistance is driven by the evolutionary pressure exerted by drugs on cell growth and may involve multiple mechanisms. Previously, extensive research on anti-virus and antibiotics has accumulated experience for understanding the mechanism of resistance. For example, resistance to antiviral drugs is usually caused by mutations in the target protein, which can prevent drug binding. Because the virus population consists of a series of related genotypes (also called virus quasispecies or groups), the selection of resistant viruses can happen quickly due to the high mutation rate during replication. In bacteria, the horizontal gene transfer of bacteria can promote the modification of drugs and make them ineffective by selecting genetic elements (for example, β-lactamase hydrolyzes β-lactam antibiotics), thereby generating acquired drug resistance.
In addition to the above mechanisms, in tumor cells, mechanisms for promoting drug resistance also include reducing the abundance of cellular drugs by up-regulating exogenous pathways that promote drug metabolism, and increasing the amount of non-specific multidrug resistance (MDR). Gene expression, etc. In summary, the mechanism of drug resistance in patients may be the composite result of multiple factors. New strategies are urgently needed to solve the emergence of drug resistance and develop long-term effective treatment methods. However, through medicinal chemistry and chemical biology The innovations will help solve the drug resistance problem.
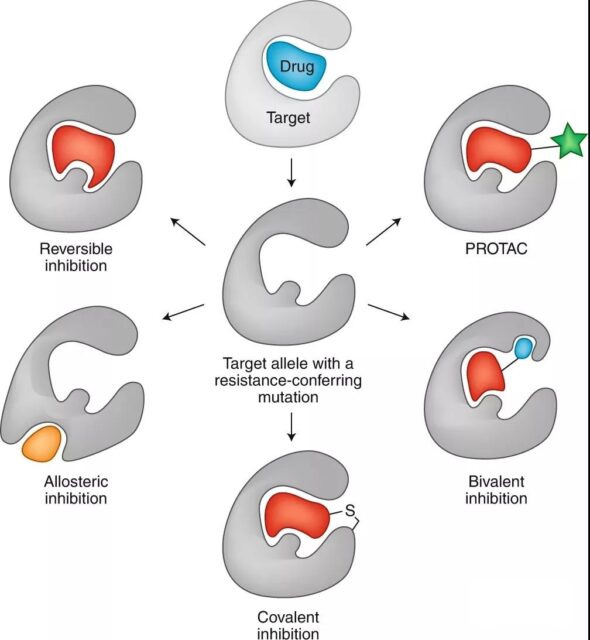
1. Design inhibitors with different binding modes
The emergence of anti-cancer small molecule drug resistance often comes from gene mutations encoding target proteins (such as BCR-ABL, EGFR or ALK), thereby blocking or reducing the binding of small molecule drugs to the original site of the target. An important example of drug resistance in this type of cancer cell is the mutation of the gatekeeper residue at the active site of the kinase, which prevents the binding of drugs acting on that site.
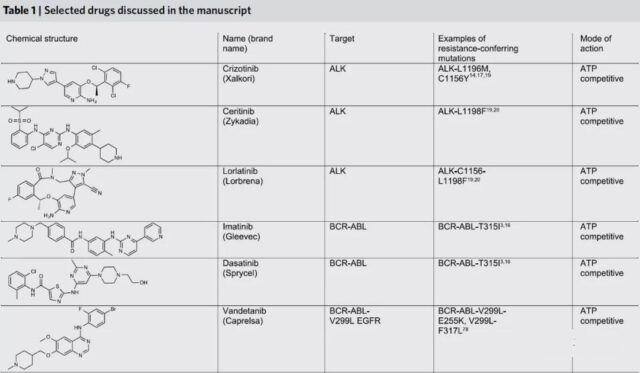
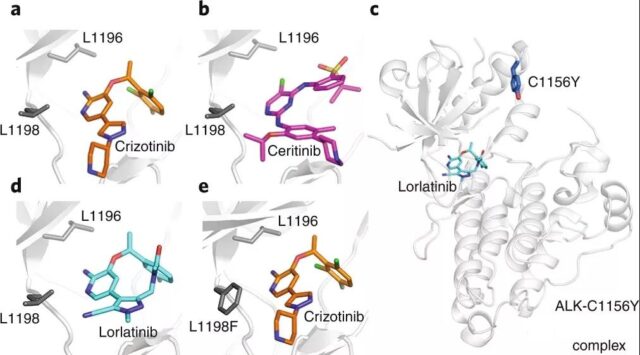
For example, in BCR-ABL-driven leukemia, the T315I mutation often occurs, preventing the binding of different competitive inhibitors that target the active site of ABL1 kinase, such as imatinib (imatinib) or dasatinib (dasatinib). Similarly, a mutation in ALK1, L1196M, can make crizotinib resistant.
Researchers overcome this type of resistance by discovering molecules with different binding modes at the same site. For example, the three generations of ALK inhibitors developed subsequently all bind to the active site of ALK, but they can achieve selectivity and efficacy by interacting with specific residues of the active site through different binding modes, thereby overcoming drug resistance.
Combining preclinical analyses of resistance and compound testing (Combining preclinical analyses of resistance and compound testing) on patients is currently a more successful method to predict and overcome resistance. As the first-generation ATP competitive inhibitors, entrectinib and larotrectinib are used to treat cancers driven by NTRK1-3 gene lesions encoding tropomyosin receptor kinases (TrkA, TrkB and TrkC). Like other molecular targeted therapies, these drugs quickly developed acquired drug resistance during use.
The drug resistance analysis of patient tumor samples after cell culture showed that the drug resistance of larotrectinib and entrectinib mainly comes from the loop exposed to the solution in front of the kinase nucleotide binding pocket (for example, G595R in TrkA and G667C and G623R in TrkC) and Caused by a mutation on the DFG motif (for example, G667C in TrkA).
Therefore, the researchers carried out compound screening for these mutant alleles and found that a series of 13-membered ring molecules based on the larorectinib backbone can effectively block the activity of the Trk alleles against larorectinib and entrectinib. One of these molecules, LOXO-195, has quickly entered clinical trials. In the treatment of patients who are resistant to larorectinib, LOXO-195 can cause tumor regression in most patients, indicating that preclinical resistance analysis is valuable for designing drug discovery that improves efficacy.
2. Design a covalent inhibitor targeting cysteine
Covalent inhibitors are another chemical strategy to overcome resistance. There are electrophilic groups (such as acrylamide, epoxy compounds or α-halocarbonyl) in these inhibitors, which can form covalents with nucleophilic residues in the site (such as cysteine or lysine) key. It is known that the active sites of many drug targets contain natural cysteine that can be used for inhibitor design.
For example, when gefitinib (gefitinib) or erlotinib (erlotinib) is used for long-term treatment of EGFR-driven cancers, It will cause the patient to develop EGFR-T790M mutation and thus become resistant. Although the mutation does not prevent the binding of these drugs at the active site, their binding affinity to ATP substrates increases, which is believed to be the cause of drug resistance.
In order to solve this problem, the researchers found that EGFR has a cysteine residue at the entrance of the ATP binding site. The electrophilic compound (C797) that forms a covalent bond with this residue can effectively block EGFR-T790M. For example, covalent inhibitors such as osimertinib or WZ4002.
Compared with reversible inhibitors, covalent inhibitors can significantly increase the occupancy rate of the inhibitor at the site, and the interaction with the methionine side chain of T790M can also help reduce the production of non-cancer cells due to the inhibition of wild-type EGFR The undesirable side effects. These developments indicate that the design of covalent inhibitors targeting the natural cysteine of the protein active site can overcome the resistance to targeted anticancer drugs.
3. Design inhibitors for allosteric binding sites
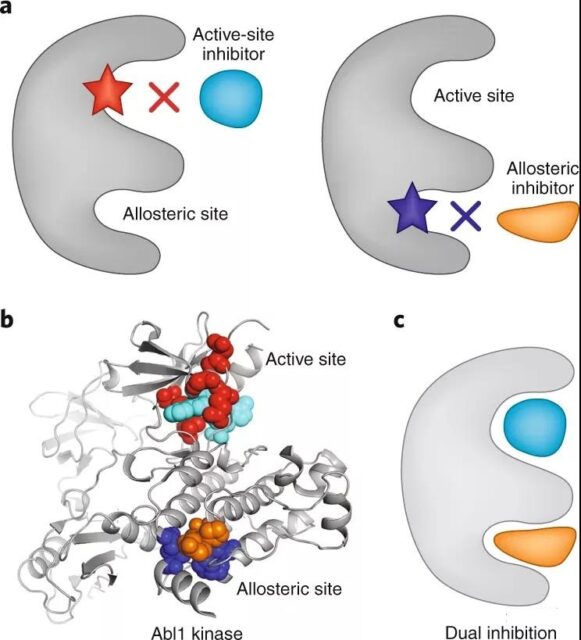
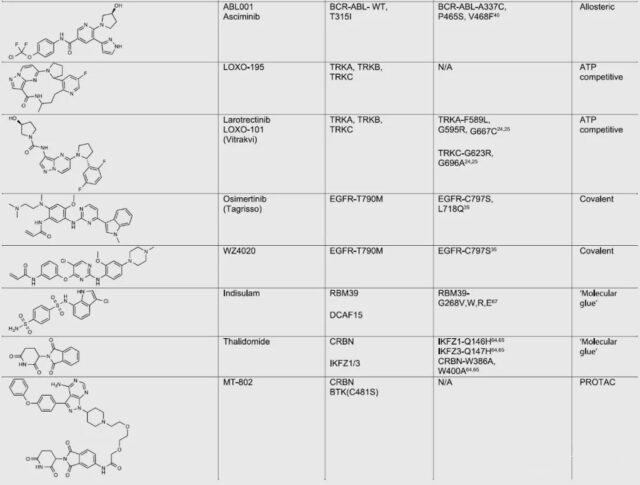
Although the drug resistance can be partially alleviated, the effectiveness of the aforementioned ATP-competitive inhibitors or covalent inhibitors may still be weakened by mutations that occur during prolonged drug treatment. A more advantageous strategy is to design inhibitors for allosteric sites (or pockets) of inactive sites in the protein. Compared with ATP binding sites, these pockets are less conservative, and inhibitors that target them may have higher selectivity and additional beneficial properties (for example, longer target residence time). For example, allosteric inhibitors against the myristic acid pocket of Abl1 kinase can effectively block kinase activity. In addition, allosteric inhibitors targeting the myristic acid pocket, such as ABL001 (asciminib), are also effective for active site mutation-resistant tumors. Therefore, drug development aimed at protein allosteric sites is one of the possible strategies to finally overcome drug resistance.
Of course, mutations may also appear in allosteric pockets and lead to resistance to these allosteric inhibitors. Studies on acquired resistance to axitinib have shown that mutations in the myristic acid pocket of Abl1 kinase (the inhibitor’s intended binding site) can develop resistance and prevent drug binding. In this case, mutations that block asciminib binding are very different from those that develop resistance to active site inhibitors. The allosteric pocket mutant BCR-ABL genotype does not significantly change the kinase activity, and can be blocked by active site inhibitors such as imatinib or dasatinib. Clinical ongoing studies have shown that combining allosteric and active site inhibitors to block BCR-ABL in cells can effectively reduce or even prevent the acquisition of resistance. Further research will determine whether this combination of drugs can also limit the development of resistance to BCR-ABL drugs in patients.
4. design compounds that can bind to multiple sites
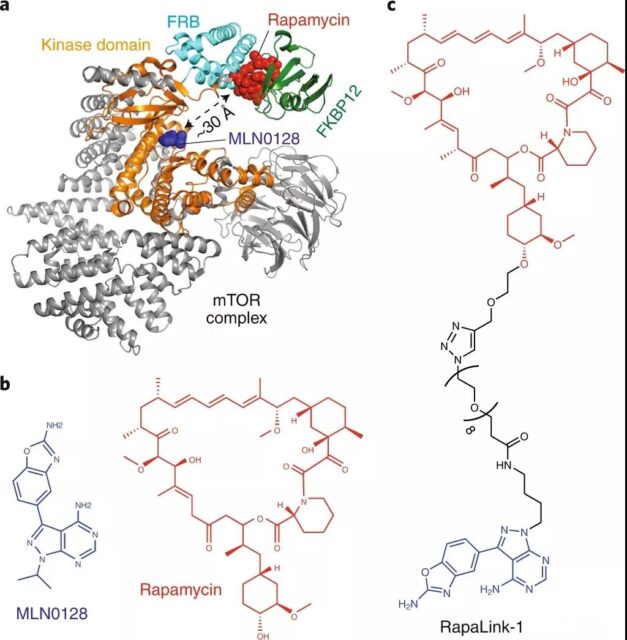
The PI3K–Akt-mTOR pathway is often overactivated in human cancers, and mTOR is one of the targets of drug development. In clinical trials of mTOR inhibitors, both the rapalogs on the rapamycin binding site or the TOR kinase inhibitors (TOR Kinase inhibitors, TORKis) on the ATP binding site induced drug resistance at the corresponding site. For example, mutations in the FRB domain (FKBP12-rapamycin-binding domain) (such as A2034V or F2108L) can prevent the binding of inhibitors such as rapalog, thereby making patients resistant to everolimus (everolimus). In addition, the M2327I mutation in the kinase domain ~ 15Å far from the ATP binding site does not prevent the binding of the compound, but it can develop resistance to the TOR kinase inhibitor AZD8055.
Recently, in order to solve the problem of resistance of these two types of mTOR inhibitors, researchers have designed a bivalent compound called RapaLink-1. By adding a linking chain between rapalog and TORKi, the compound can be combined with rapamycin at the same time. The site interacts with the ATP binding site. Cell studies have shown that RapaLink-1 can inhibit proliferation by blocking mTOR signaling, and suggests that the compound binds to two pockets of the target. Interestingly, RapaLink-1 also has an inhibitory effect on the mutations of mTOR in FRB and kinase domains, which overcomes the resistance of mutations to rapalogs, TORKi and their combinations. Bivalent compounds like RapaLink-1 can also improve the selectivity for the selected target, and similar methods also bring positive ideas for the design of dual-site drugs for other targets.
5. Design PROTACSs to overcome resistance
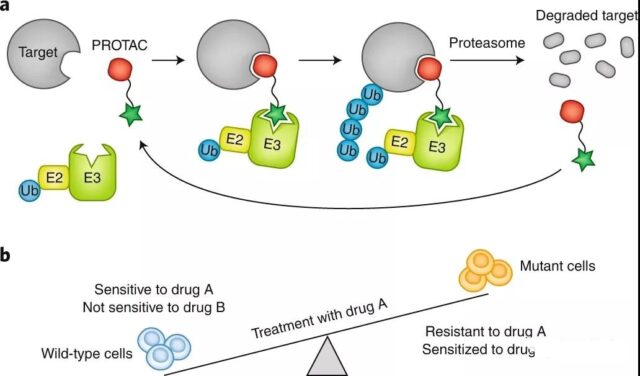
PROTACs refer to a class of bifunctional molecules that use targeting to induce protein degradation, which can inhibit the function of the target, and can also target the drug-resistant mutation of the target in cancer. It is considered to be a promising strategy. The PROTAC molecular structure consists of three parts: “target protein ligand-linker-ligase ligand”. The ligase ligand can recruit E3 ubiquitin ligase, such as von Hippel-Lindau (VHL) or cereblon (CRBN). Enzyme. The ligase ligand in the PROTAC molecule is connected to the ligand that interacts with the target protein through a covalent linker.
Through the ligands at both ends, the PROTAC molecule, the target protein and the E3 ligase can lead to the formation of a ternary complex, which promotes the ubiquitination of the target protein by the E3 ligase and the degradation of the ubiquitinated target protein by the proteasome. However, in order to bind two different proteins, the PROTAC concentration must be adjusted precisely to avoid the formation of ineffective dimers with the target protein or E3 ligase at high concentrations, thereby preventing the formation of a ternary complex (hook effect). In addition to complex pharmacology, it is also difficult to develop optimal linkers for PROTACs.
Interestingly, when the degraded ubiquitinated protein releases the PROTAC molecule, the PROTAC molecule can bind to another molecule of the target protein and repeat the degradation cycle. This mode of action is different from common inhibitor-target binding occupancy-driven pharmacology, which may require high-dose inhibitors to effectively block the activity of the target. Compared with reversible inhibition, another advantage of targeted degradation is that it can block the target for a longer time, because its function and activity can only be restored by the re-synthesis of the protein.
The PROTAC method has been proven to be suitable for the degradation of a variety of cancer proteins (for example, BCR-ABL, EGFR or BTK). Recent studies have also shown that PROTACs can overcome drug resistance caused by mutations in these targets.
For example, BTK resistance mutations are usually caused by the substitution of serine (C481S) for cysteine residues in the active site, which reduces the binding of the covalent inhibitor ibrutinib. PROTACs based on ibrutinib (such as MT-802), despite their low potency, can still bind to mutant proteins and induce the degradation of resistant BTK.
This suggests that the use of the PROTAC method may be able to target the drug-resistant alleles of anti-tumor drugs. In the case where the drug-resistant mutation on the target completely prevents the binding of the original molecule, it may be necessary to target the allosteric site PROTACs on the protein to provide another degradation mode (for example, the allosteric site of BCR-ABL).
6. Overcoming drug resistance through the use of drug combinations
Combination therapy is a mature clinical method for cancer treatment, and it can also help resolve emerging drug resistance. Mixed therapy using multiple chemotherapeutic drugs has become a successful cancer treatment method because the drug combination has a synergistic effect and provides more effective anti-cancer results than a single drug. Use the principle of non-overlapping resistance spectrum and synthetic lethality to help overcome resistance.
Drug resistance is usually caused by genetic changes in the drug target (for example, point mutations, deletions, or errors in expression regulation). To develop resistance to drug combinations that target different proteins or binding sites requires multiple changes. . However, given that multiple genetic changes (such as single point mutations) are less likely to occur simultaneously in a single cell, a combination of drugs with non-overlapping drug-resistant mutation profiles (for example, for “normal-alternative” on proteins) is used. Combination of sites or targeting different protein targets) may be a powerful strategy to limit and prevent the emergence of drug resistance
Future outlook
Drug resistance is a major scientific issue in the development of targeted drugs. With the emergence of some innovative technologies in medicinal chemistry and chemical biology, the development of drug development strategies that have better curative effects and respond to the emergence of drug resistance in advance can be carried out.
In these methods, simultaneous targeting of the orthosteric active site and allosteric site on a protein has been proven to reduce the emergence of drug resistance, because multiple mutations that prevent compound binding are less likely to occur, which is a solution to resistance. One of the most promising options for medicine.
At the same time, advances in computational drug design/machine learning may identify different drug combinations that have the potential to overcome resistance. The development of PROTACs and multi-site binding molecules will also help develop drugs that can reduce or even prevent the emergence of resistance. These new technologies will help achieve the hope of targeted cancer cures.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



