Advancements and Challenges in CAR-T Therapy for T-Cell Malignant Tumors
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Advancements and Challenges in CAR-T Therapy for T-Cell Malignant Tumors
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Advancements and Challenges in CAR-T Therapy for T-Cell Malignant Tumors
Chimeric Antigen Receptor T-cell (CAR-T) therapy has been widely used in the treatment of various types of relapsed/refractory (R/R) B-cell malignant tumors.
Currently, the U.S. Food and Drug Administration (FDA) has approved five CAR-T products.
Tisagenlecleucel, Axicabtagene ciloleucel, and Lisocabtagene Maraleucel are CAR-T products for treating R/R diffuse large B-cell lymphoma (DLBCL).
Brexucabtagene autoleucel is used for mantle cell lymphoma (MCL). Additionally, Idecabtagene vicleucel, the latest CAR-T product targeting BCMA, has FDA approval for certain multiple myeloma (MM) cases.
Despite the clinical success in B-cell malignancies, CAR-T therapy still faces challenges in certain types of hematologic malignancies and solid tumors.
T-cell malignant tumors, including T-cell acute lymphoblastic leukemia (T-ALL), T-cell large granular lymphocytic leukemia (LGL), adult T-cell leukemia/lymphoma (ATL or ATLL), T-cell prolymphocytic leukemia (T-PLL), and peripheral T-cell lymphomas (PTCLs), remain an area where CAR-T therapy has not yielded favorable results.
Compared to B-cell malignancies, first-line cancer treatments (including chemotherapy) for T-cell malignant tumors have limited clinical responses, leading to poor prognoses for these patients.
Given the success of CAR-T therapy in B-cell malignancies, there is an expectation that CAR-T therapy could also improve clinical outcomes for T-cell malignant tumor patients.
However, CAR-T therapy for T-cell tumors remains challenging, with issues such as a lack of T-cell tumor-specific CAR-T targets and challenges related to self-killing, T-cell regeneration disorders, and contamination with malignant T cells during autologous CAR-T production requiring further research.
Target Antigens, Self-Killing, and Solutions
CD3
CD3 is a pan-T surface antigen that forms a complex with the T-cell receptor (TCR), leading to T-cell activation upon recognition of target antigens. CD3 is present on all mature T cells, making it a favorable target for immunotherapy of T-cell malignant tumors. However, using CD3 as a CAR-T target in T-cell malignant tumors may lead to adverse reactions due to self-killing.
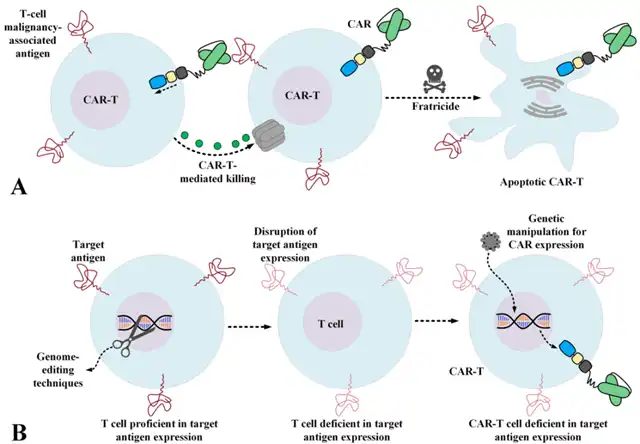
Therefore, efforts have been made to generate CAR-T cells resistant to self-killing or to use other types of effector cells, such as NK cells, which may eliminate the above problems to some extent. Genome editing methods provide various ways to disrupt antigens that cause self-killing, resulting in CAR-T cells with specific and significant anti-tumor activity against primary T cells and pediatric T-ALL samples.
In addition, other researchers have designed third-generation CD3-targeted CARs and expressed them in the NK cell line NK-92. Compared to T cells, NK cells lack CD3 expression, have a shorter lifespan, and exhibit anti-tumor activity against CD3-expressing PTCL samples and various T-cell leukemia cell lines.
CD5
CD5 is a transmembrane glycoprotein involved in the survival of human lymphocytes and acts as a negative regulator of TCR signaling. CD5 is normally expressed only in thymic cells, peripheral T lymphocytes, and a B lymphocyte subset called B-1a cells. Abnormal expression of CD5 has been detected in several T-cell malignant tumors, including T-ALL and PTCL.
In 2015, Mamonkin and colleagues designed CD5 redirected CAR-T cells, reporting that these cells underwent partial and transient self-killing but had specific anti-tumor activity. Researchers also expressed third-generation CARs in the NK-92 cell line and demonstrated that these CAR-NKs, lacking CD3 expression compared to T cells, showed anti-tumor activity against human T-ALL, PTCL, and primary CD5+ cells. Furthermore, using co-stimulatory domains 4-1BB or 2B4 significantly enhanced anti-tumor activity.
CD7
CD7 is a transmembrane glycoprotein from the Ig superfamily, usually expressed on NK cells and T lymphocytes. Studies indicate that CD7 is overexpressed in a high proportion of T-ALL and T-cell lymphoma cases.
However, targeting CD7 with CAR-T production has been challenging because T cells themselves express CD7, which may lead to self-killing and disrupt the amplification of CAR-T products. In 2017, GomesSilva and colleagues used CRISPR-Cas9 to disrupt CD7 expression in T cells, then converted them into CAR-T cells. This method not only had no negative impact on the anti-tumor function of these CAR-T cells but also increased their amplification efficiency. These CAR-T cells showed unique anti-tumor activity against various CD7-expressing cell lines and T-ALL.
In the first human clinical trial of CD7-targeted CAR-T (NCT04004637), researchers used CD7-specific nanobodies as the targeting domain for CAR-T. Additionally, they used intelligent technology to prevent CD7 surface expression and subsequent self-killing by retaining the antigen in the endoplasmic reticulum and/or Golgi apparatus. Results showed robust amplification and acceptable persistence in two patients (66%), and both patients reported minimal residual disease (MRD) negativity within less than a month, with no detectable abnormal T cells. However, varying levels of cytokine release syndrome (CRS) and increased IL-6 levels were observed in all patients. Other clinical trials (NCT04033302 and NCT03690011) are also testing the applicability and efficacy of CD7-targeted CAR-T in various types of T-cell malignant tumors.
CD1a
CD1a is a cell surface antigen present on cortical T-ALL cells, with specific expression observed in developing cortical thymic cells. T cells and CD34+ hematopoietic progenitor cells do not show CD1a expression. The specificity of CD1a makes it a suitable target antigen, minimizing the potential for off-target toxicity when targeting non-tumor cells.
A research group explored CAR-T targeting CD1a and found that these cells, in addition to being self-killing resistant, exhibited strong anti-tumor activity against primary cells from CD1a-expressing T-ALL cell lines and cortical T-ALL samples. Moreover, in in vivo assessments using patient-derived xenograft (PDX) models, these effector cells showed persistent and substantial anti-tumor activity after administration.
CD4
Several studies have investigated CD4-targeted CAR-T. In 2016, Pinz and colleagues studied third-generation CD8+CD4-targeted CAR-T and reported that these cells displayed unique anti-tumor activity against CD4-expressing cell lines and patient-derived PTCL cell samples while retaining their memory stem cell-like phenotype.
In 2017, Pinz and colleagues generated third-generation CD4-targeted CAR-NK cells using the NK-92 cell line. In xenograft models, these CD4-targeted
CAR-NK cells effectively infiltrated the tumor site, causing regression of CD4-expressing PTCL cell lines, patient-derived T-ALL cells, and T-PLL.
Another study used CD4-specific nanobodies as the targeting domain and performed clinical trials on CD4-targeted CAR-T cells in T-ALL patients (NCT04071366). The results showed stable amplification, effective migration to tumor sites, and robust anti-tumor activity in all patients. Nevertheless, the trial was halted due to the emergence of graft-versus-host disease (GVHD) and Epstein-Barr virus (EBV) reactivation. In another trial, CD4-targeted CAR-T cells were genetically modified to express a suicide gene that would eliminate CAR-T cells in response to GVHD. This approach, combined with administration of a CD4 monoclonal antibody (Mab), demonstrated acceptable safety and efficacy in clinical trials (NCT03690011).
CD30
CD30, also known as TNFRSF8, is expressed after T cells and B cells receive activation signals stimulated by target antigens. CD30 is also expressed in various T-cell malignancies, including T-ALL and anaplastic large cell lymphoma ( ALCL ).
Currently, CD30-targeting CAR-T is being studied in different stages of clinical trials. In 2017, a phase I dose-escalation clinical trial ( NCT01316146 ) was reported in which 7 patients with R/R HL and 2 ALCL received CD30-targeted second-generation CAR-T cells. No toxicity related to CAR-T was reported in the study. In addition, among the 7 HL patients in this study, there were 2 cases of CR ( one case lasted more than 2.5 years and the other case lasted about 2 years ) and 3 cases SD. In addition, 1 of 2 ALCL patients experienced a CR that lasted 9 months. In the same year, a report from another clinical trial ( NCT02259556 ) involving 18 patients with progressive R/R HL showed that only 2 patients experienced severe toxicity and the remaining patients tolerated CAR-T infusion well. In terms of efficacy, 7 patients had partial response and 6 patients had stable condition.
CD37
CD37 is a tetrapeptide leukocyte-specific surface antigen expressed on mature normal and transformed B cells. CD37 also has a regulatory role in T cell proliferation. CTCL and PTCL are T-cell malignancies in which CD37 expression has been detected.
In 2018, Scarfò et al. studied CD37-targeted CAR-T and reported that these cells mediated target antigen-dependent activation, cytokine secretion, and tumoricidal activity against T-cell lymphoma in vitro without any apparent autologous effects. Signs of cannibalism. The in vitro evaluation of this study involved cell lines such as Hut78 and Fedp as well as PTCL patient-derived cell samples, all of which have varying levels of CD37 expression. However, the study also noted that because not all PTCL cell lines or patient-derived samples are CD37+, screening for the expression of this antigen may be necessary in future preclinical and clinical studies.
CCR4
CCR4 is a chemokine receptor expressed by normal T cell subsets, including regulatory T cells ( Treg ), Th2 and Th17 cells. Furthermore, overexpression of this chemokine receptor was detected in malignant T cells from patients with ATLL, PTCL, and CTCL, including mycosis fungoides ( MF ) and Sézary syndrome ( SS ).
In 2017, Perera et al. studied that CCR4-targeted CAR-T could mediate an effective target antigen-dependent anti-tumor response against patient-derived tumor cell lines expressing CCR4. In addition, tumoricidal activity has been demonstrated in xenograft models of adult T-cell leukemia. However, CCR4 expression on normal T cell subsets may lead to unexpected toxicities, which may require further in-depth evaluation.
TRBC1 and TRBC2
Most PTCL is TCR+. TCR has α chain and β chain. T cell receptor β constant region 1 ( TRBC1 ) and T cell receptor β constant region 2 ( TRBC2 ) genes are responsible for the expression of TCR β chain constant region. In a normal T cell population, there is a mixture of TRBC1-expressing cells and TRBC2-expressing cells. Instead, the entire malignant T cell population will express only TRBC1 or TRBC2. Thus targeting TRBC1 ( in the case of TRBC1-expressing T-cell malignancies ) or targeting TRBC2 ( in the case of TRBC2-expressing T-cell malignancies ) can eliminate tumor T cells and a subset of normal T cells expressing the target beta chain constant region , but will not produce any tumoricidal effect on most normal T cells.
Maciocia et al. studied TRBC1-targeted CAR-T cells that mediated specific antitumor responses against malignant TRBC1+ but not TRBC2+ cells in vitro. An ongoing clinical trial ( NCT03590574 ) is testing a TRBC1-targeted CAR-T therapy called AUTO4 for TRBC1+ T-cell non-Hodgkin lymphoma ( T-NHL ), PTCL, angioimmunoblastic T-cell lymphoma safety and efficacy in patients with tumours, AITL and ALCL.
T cell aplasia and its solutions
T-cell aplasia is the result of CAR-T targeting of normal T cells, and T-cell aplasia significantly increases the risk of various life-threatening infections. Therefore, preventing this adverse event is necessary for successful CAR-T therapy in patients with T-cell malignancies.
The development of T-cell aplasia can be prevented through various strategies. One strategy is to select target antigens. Targeting antigens that are missing on normal T cells or that are expressed on some T cells may keep at least a certain proportion of normal T cells intact during CAR-T therapy. Another strategy is to use CAR-T with limited or controllable longevity or activity, whose limited anti-tumor effects help prevent the occurrence of T-cell aplasia. In addition, allogeneic hematopoietic stem cell transplantation ( HSCT ) after CAR-T therapy may also be another option to alleviate CAR-T-related T cell aplasia.
Another applicable strategy to prevent T cell aplasia is to equip CART with a safety switch (also known as a suicide switch) that would allow it to control adoptively transferred T cells after administration to the patient. So far, different safety switch platforms have been introduced, including metabolic switches, monoclonal antibody-dependent switches, and inducible caspase ( iCasp ) switches. Currently, multiple research teams are currently investigating the suitability of these safety switches in clinical settings ( NCT02028455, NCT03016377, or NCT01815749 ).
Using viral vectors to transduce CAR-T can stably express CAR in transduced T cells. These transduced T cells can expand in vivo while maintaining CAR expression, and this open duration of CAR expression may result in on-target non-tumor toxicities such as T cell aplasia. CAR-T generated using CAR mRNA electroporated into T cells exhibit limited persistence after administration. Two clinical trials ( NCT02277522 and NCT02624258 ) investigated the use of non-viral mRNA electroporated CD19-targeted CAR T in patients with R/R HL. According to the report, no reactions and signs of serious toxicity were observed during these trials. Considering that this method may help reduce the targeted non-tumor toxicity of CAR-T therapy in patients with T-cell malignancies, it is speculated that continuous CAR-T administration may be required to obtain a stable and reliable anti-tumor response.
Contamination of malignant T cells and their solutions
Generating autologous T cells from patients with T cell malignancies is challenging because during the T cell isolation process, both normal and malignant T cells are isolated. It is more likely to occur when patients with T-cell malignancies ( especially T-cell leukemias with high numbers of circulating malignant T cells ) produce autologous CAR-T.
In this case, generating allogeneic CAR-T from healthy third-party donors may be considered a suitable solution. However, allogeneic CAR-Ts also have their problems, for example, they may mediate life-threatening GvHD, and they may be rapidly attacked and eliminated by the host’s immune system. Both of these obstacles significantly weaken the tumoricidal activity of CAR-Ts. In this regard, researchers have developed allogeneic CAR-T to address the above limitations using various strategies. Importantly, allogeneic CAR-Ts have shorter in vivo persistence after infusion into patients compared with autologous CAR-Ts, a property that helps prevent T cell aplasia.
α- and β-knockout allogeneic CAR-T targeting CS1 and CD22 are currently in clinical studies for the treatment of MM ( NCT04142619 ) and B-ALL ( NCT04150497 ) respectively. Use of these methods can help generate off-the-shelf allogeneic CAR-Ts that can be safely used clinically to treat patients with T-cell malignancies.
Another strategy is to use multivirus-specific T ( multiVST ) cells as effector cells for CAR expression. These cells are genetically engineered to lack expression of the CAR target antigen, thereby rendering them resistant to cannibalism. A clinical study ( NCT01570283 ) demonstrates that multiplexed VST cells are safe and capable of mediating clinical responses in immunocompromised allograft recipients. Using these T cells may be a suitable method to generate off-the-shelf CAR-T cells.
γδ T cells make up 1% to 5% of circulating lymphocytes, which are ubiquitous lymphocytes in the skin, reproductive system, and intestines. This property of γδ T cells is very beneficial in adoptive cell therapy because αβ T cells have difficulty entering these sites. In addition, γδ T cells express chemokine receptors that interact with chemokines secreted by tumor cells. This property helps γδ T cells migrate more easily to tumor sites. Therefore, these properties make γδ T cells CAR-expressing and allogeneic. A suitable platform for adoptive cell therapy.
Furthermore, NK cells can serve as reliable effector cells for CAR expression to reduce or eliminate the possibility of cannibalism. On the other hand, NK-expressing CARs are not as durable as traditional CAR-T, a property that can minimize the possibility of T cell aplasia. Most importantly, NK-based CAR-expressing effector cells do not possess TCR and do not cause GvHD.
Summary
CAR-T therapy for T-cell malignancies is one of the most complex and challenging areas in cancer immunotherapy.
Compared with CAR-T therapy for B-cell malignancies, this field is fraught with difficulties due to issues of antigen selection, cannibalism, T-cell aplasia, and malignant T-cell contamination.
However, people have developed a variety of methods to solve these problems, including gene editing.
The application of allogeneic CAR-T and CAR-NK will give us the opportunity to make breakthroughs in cell therapy of T-cell malignancies.
At the same time, it is also needed More preclinical and clinical data are needed to validate the safety and efficacy of these approaches.
Advancements and Challenges in CAR-T Therapy for T-Cell Malignant Tumors
References:
1.CAR-T cell therapy in T-cellmalignancies: Is success a low-hanging fruit? Stem Cell Res Ther. 2021;12: 527.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



