Tumor therapy targeting DNA repair pathways
- Why Lecanemab’s Adoption Faces an Uphill Battle in US?
- Yogurt and High LDL Cholesterol: Can You Still Enjoy It?
- WHO Releases Global Influenza Vaccine Market Study in 2024
- HIV Infections Linked to Unlicensed Spa’s Vampire Facial Treatments
- A Single US$2.15-Million Injection to Block 90% of Cancer Cell Formation
- WIV: Prevention of New Disease X and Investigation of the Origin of COVID-19
Tumor therapy targeting DNA repair pathways
- Red Yeast Rice Scare Grips Japan: Over 114 Hospitalized and 5 Deaths
- Long COVID Brain Fog: Blood-Brain Barrier Damage and Persistent Inflammation
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Tumor therapy targeting DNA repair pathways.
Foreword
Over the past few decades, the understanding of DNA damage response ( DDR ) pathways has grown, broadening the therapeutic landscape of oncology.
It is increasingly clear that genomic instability in cells caused by defects in the DNA damage response contributes to the development of cancer.
On the other hand, these deficiencies can also serve as a therapeutic opportunity.
A growing number of DDR-targeted drugs have rapidly expanded to inhibitors of multiple members of the DDR pathway, including PARP, ATM, ATR, CHK1, WEE1, and DNA-PK.
Currently, inhibitors of these DDR components, some of which are under clinical investigation.
In addition, emerging evidence for the sensitivity of DDR inhibitors to conventional cancer treatments, as well as the correlation between DDR pathways and immune checkpoint inhibitor (ICI) responses, are promoting DDR inhibitor-based combination therapy.
Drugs targeting DNA repair pathways are showing an increasing role in the field of tumor therapy.
DNA damage and DNA damage response
To maintain genome integrity, complex DNA repair systems are employed to combat various forms of DNA damage, and these mechanisms are known as the DNA damage response.
The DDR pathway is divided into three functionally intertwined parts: sensors that detect DNA damage, signal converters that trigger signaling cascades, and effectors that hinder DNA repair.
These pathways are not mutually exclusive processes, but coordinate with each other to form precise regulatory networks for DNA repair.
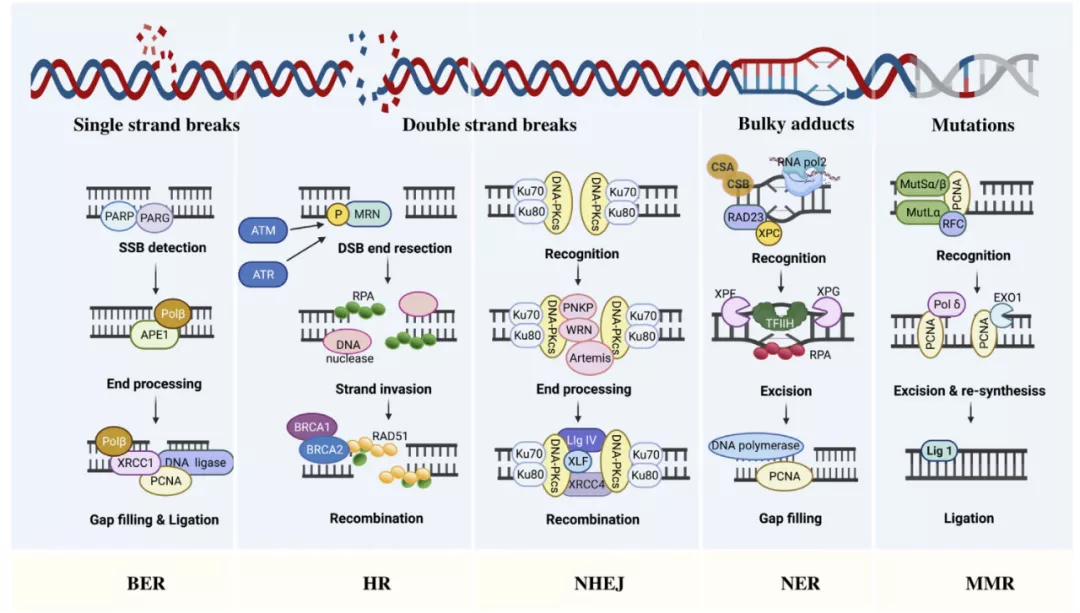
Base Excision Repair (BER) and Nucleotide Excision Repair (NER)
The genomes of all organisms are constantly undergoing subtle changes as a result of endogenously produced various genotoxic agents such as reactive oxygen species ( ROS ), ionizing radiation, and environmental damage such as alkylating agents. Most subtle changes in DNA, such as single-strand breaks ( SSBs ), are repaired through the BER pathway.
BER is first initiated by damaged bases, then the bases are excised and replaced with newly synthesized DNA.
Then, apurinic/apyrimidinic endonuclease ( APE ) cleaves the AP site to form a 3′OH terminus at the damaged site.
Finally, DNA polymerase and DNA ligase are recruited at the nucleotide gap created by removing the damaged base, thereby closing the gap. BER is responsible for repairing small lesions, while larger double-strand breaks ( DSBs ) that deform the DNA helix structure require NER pathway repair.
The NER machinery involves a key protein, excision repair cross-complementary protein 1 ( ERCC1 ), which clears DNA near the breakpoint and then replaces it with normal DNA replication.
Homologous recombination ( HR ) and non-homologous end joining (NHEJ)
In mammalian cells, HR and NHEJ are the two main pathways for DSB repair.
Since homologous sister chromatids are templates required for new DNA synthesis, the HR pathway can repair DSBs in the S/G2 cell cycle phase, while NHEJ is active in all cell cycle phases except M phase.
HR analyzes homologous sequences from other parts of the genome to collect information about the loss of break sites.
HR first excises the broken end, and then forms Rad51 nucleoprotein filaments through Brca2 and Rad51, starts to retrieve homologous sequences, and promotes the formation of linking molecules between the broken DNA and the homologous template to complete the repair.
NHEJ is a bit simpler than HR, directly rejoining the broken ends together.
The essential factor required for NHEJ is a heterodimer consisting of Ku70/Ku80 and the catalytic subunit of DNA-dependent protein kinase ( DNA-PKcs ) , which recognize DSBs and promote downstream signaling factors of NHEJ, such as XRCC4, XLF and DNA Ligase IV.
Although the NHEJ mechanism is simpler, it sometimes leads to rearrangements, and HR is not thought to produce errors.
In addition to HR and NHEJ, one DSB repair pathway has a similar mechanism to these two major DSB repair pathways, but is genetically distinct, called the alternative end joining ( a-EJ ) pathway.
The a-EJ pathway can either share a similar initiation process with HR or constitute a homologous template-free DNA end-linking factor like NHEJ.
Currently, more and more studies have begun to focus on the a-EJ pathway as a potential therapeutic target for cancer cells with impaired NHEJ or HR activity.
Mismatch Repair (MMR)
In addition to damage produced by cells exposed to genotoxins, DNA damage may also result from abnormal DNA processing.
The DNA repair pathway for replication-related errors is called MMR. During DNA synthesis, MMR corrects nucleotide misintegrations, thereby preventing permanent DNA changes in dividing cells.
Therefore, MMR deficiency caused by genetic mutation or epigenetic silencing may lead to an increased incidence of spontaneous mutations, which are often associated with hereditary and sporadic cancers.
Translesion synthesis and template replacement
DNA damage tolerance ( DDT ) acts as an essential bypass mechanism for repairing replication stalled DNA damage, allowing DNA replication to pass through blocking elements.
Translesion synthesis ( TLS ) is one of two distinct modes of DDT that depend on the function of a particular TLS polymerase, rather than a replicative DNA polymerase, that can replicate directly across the lesion site.
The TLS mechanism is error-prone due to insufficient proofreading activity of the TLS polymerase, which increases the risk of mutation. Not surprisingly, TLS is a major source of cellular mutations.
In contrast, another mode of DDT, template displacement ( TS ), involves recombination on sister chromatids to a homologous DNA template, which is similar to the HR process and is thought to be more accurate than TLS.
The repair activities of TLS and TS begin after the replication fork, suggesting that they may occur during or after DNA replication, with TS beginning in early S phase and TLS beginning in late S phase.
Fanconi Anemia (FA) Pathway
Fanconi anemia is a rare genetic disorder caused by biallelic mutations in the Fanconi gene that affects patients with an inadequate response to DNA damage.
Fanconi anemia has been identified as a DNA repair pathway that removes barriers that impede DNA replication and transcription, known as DNA interstrand crosslinks ( ICL ).
ICLs can be formed by aldehydes during various metabolic reactions ( eg, lipid peroxidation and ethanol metabolism ) and chemotherapy ( eg, platinum ).
Whereas intrachain crosslinks are repaired through the NER pathway, ICLs are mainly repaired through the FA pathway.
Upon detection of ICL by the UHRF1 protein and the FANCM–MHF1–MHF2 complex, the FA core complex is recruited to chromatin and monoubiquitinates the substrates FANCI and FANCD2.
Ubiquitinated FANCD2-I recruits scaffold proteins for various endonucleases to complete DNA recognition and nucleotide excision at cross-links, resulting in DNA substrates suitable for recombination repair.
O6-methylguanine-DNA methyltransferase pathway
It is well known that DNA methylating agents are able to inhibit DNA methylation and generate a wide range of DNA adducts, such as O6-methylguanine ( O6MeG ) and O4-methylthymine, which can lead to base mismatches and subsequent point mutation.
Among them, O6MeG is considered to be the main source of DNA adducts induced by methylating agents, which can lead to mutagenesis and carcinogenesis.
O6MeG can be repaired in a one-step suicide reaction by O6-methylguanine-DNA methyltransferase ( also known as MGMT ).
MGMT transfers a methyl group from the damaged guanine O6 site to a cysteine residue, thereby preventing gene mutation.
Conceivably, MGMT reduces the action of alkylating agents in cancer cells, potentially leading to chemoresistance.
Methylation of the MGMT promoter impedes its transcription and can therefore be used to increase the sensitivity of cells to alkylating agents.
Mechanisms of DDR therapeutic application
The underlying mechanisms underlying the increased susceptibility of tumor cells to DNA damage compared to normal cells lie in three main aspects: defects in at least one DDR pathway, elevated replication stress, and increased endogenous DNA damage.
DDR defects
Although DDR defects have been implicated in cancer initiation and progression, defects in the DDR pathway also provide therapeutic opportunities for targeting tumor cells.
DDR-deficient tumor cells lead to enhanced genomic instability and depend on the remaining DDR pathways for survival.
Combinatorial targeting of the remaining DNA repair pathways as a therapeutic approach has given rise to a concept known as “synthetic lethality.”
The concept of synthetic lethality is based on two simultaneous loss-of-function genetic events, neither of which alone cause damage, but work together to cause cell death.
When a genetic change occurs in the DDR pathway unique to cancer cells, a second loss-of-function event caused by a DDR inhibitor is synthetically lethal to cancer cells without affecting normal cells.
Replication stress
The complex DNA replication system of eukaryotic cells is tightly regulated by various proteins in the cell cycle during cell division.
Many DNA nucleotides require precise polymerization to ensure cellular homeostasis.
Endogenous or exogenous barriers that impede or terminate replication fork progression activate conserved cellular response pathways known as replication stress.
The molecular mechanism of replication stress is the stalled progression of DNA polymerase and the subsequent uncoupling of DNA polymerization from DNA helicase.
An example of induced replication stress is G1/S cell cycle checkpoint deficiency, which may be due to loss of pRb function, loss of CDKN2A, or cyclin D1 or cyclin E amplification.
endogenous DNA damage
Some endogenous DNA damage is caused by the action of low concentrations of hydrogen and hydroxide ions at neutral pH, such as depurination and depyrimidination.
The intracellular environment presents other dangers to chromosomal DNA, the most serious of which arise from the oxidative process.
Some intermediates produced in the mitochondria, such as ROS, may leak out of the mitochondria and into the cytoplasm.
Such intermediates include superoxide ions, hydrogen peroxide and hydroxyl radicals.
In tumor metabolism, low pH, hypoxia, and high levels of ROS are prevalent in the tumor microenvironment ( TME ).
Inhibitors targeting DNA repair pathways
Current anticancer strategies exploiting DDR deficiencies have largely been addressed by developing targeted drugs that inhibit molecules during DNA repair.
Poly ADP-ribose polymerase (PARP) inhibitors
The development of PARP inhibitors is a classic example of synthetic lethality. PARP1 and PARP2 are key DDR enzymes that modify target proteins through negatively charged poly ADP-ribose ( PAR ) chains, sense DNA damage and transmit signals.
The structural change of PARP1 upon binding to damaged DNA activates its catalytic function, promoting the recruitment of DNA repair effector molecules and the structural remodeling of chromatin around DNA damage sites, and the dynamic remodeling of chromatin will largely affect DNA repair s efficiency.
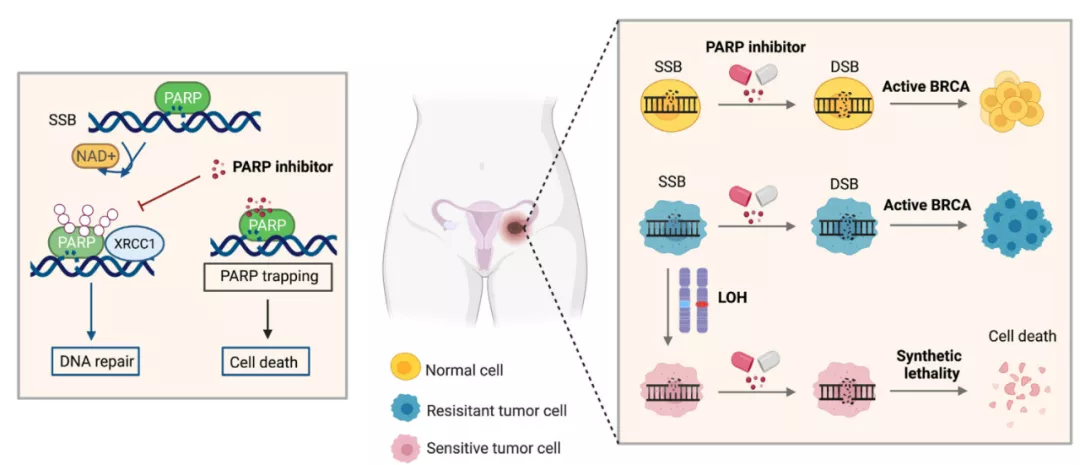
Given the critical role of PARP in promoting efficient DNA repair, PARP inhibitors can selectively kill homologous recombination-deficient tumor cells.
PARP inhibitors are a promising therapeutic strategy for BRCA-mutated tumors.
Poly ADP-ribose hydrolase (PARG) inhibitor
PARG reverses the action of PARP enzymes by hydrolyzing PAR ribose bonds after DNA damage.
Likewise, the active role of PARG in DNA replication and repair results in increased susceptibility of PARG-deficient cells to DNA-damaging agents.
Although numerous studies have demonstrated a correlation between PARP inhibitors and synthetic lethality, research on the therapeutic mechanisms of PARG inhibitors is relatively lagging.
Loss of HR proteins such as BRCA1/2 in breast cancer cells has been reported to stimulate synthetic lethality in PARG-suppressed cells, and the PARG inhibitor COH34 can induce BRCA-mutated or olaparib-resistant ovarian and breast cancer cells die.
However, conflicting results have been reported in other cancer cells. Of the six breast cancer cell lines tested, only one BRCA-deficient cell line was sensitive to the PARG inhibitor PDD00017273, while five were ineffective against PDD00017273 ( including BRCA-mutated cell lines ).
Ataxia Telangiectasia Mutated Protein (ATM) Inhibitor
The DDR signaling cascade is driven by a series of protein phosphorylations, and ATM, ATR and DNA-PKs are key kinases involved in this process.
Activated by DNA double-strand breaks, ATM is recruited to DSB sites by the MRE11-RAD50-NBS1 ( MRN ) complex.
ATM substrates include p53, CHK1 and CHK2, and their phosphorylation will result in intra-S or G2/M cell cycle arrest.
Although ATM plays a typical role in a variety of molecular processes such as DNA repair, it also has atypical functions, including spliceosome replacement.
ATM is considered a tumor suppressor, and ATM deficiency or mutation is common in solid tumors and B-cell lymphomas.
ATM mutations may lead to ataxia telangiectasia, a neurodegenerative disease that predisposes to cancer. Several ATM inhibitors are currently under investigation for cancer treatment.
Ataxia telangiectasia and Rad3-related protein (ATR) inhibitors
Unlike ATM, which is triggered by DSB, ATR is activated and recruited by replication protein A ( RPA )-wrapped single-stranded DNA.
Single-stranded DNA can be produced by nucleolytic processing of DSBs and uncoupling of replicating DNA helicases from DNA polymerases.
Intracellular ATR signaling involves phosphorylation of a range of downstream molecules, triggering a wide range of responses, including blockade of cell cycle checkpoints, DDR, and apoptosis.
Compared with other DDR proteins such as PARP, the development of ATR inhibitors is relatively lagging.
Reasons may include the large size of the ATR molecule and the lack of understanding of its crystal structure.
Furthermore, its highly homologous active sites in all PIKKs and the requirement for co-activating proteins further limit its drug design.
CHK1 inhibitors
The checkpoint kinase CHK1 is actively involved in the DNA damage response initiated by ATR and ATM by phosphorylating and recruiting a series of regulatory proteins.
CHK1 regulates the S-phase checkpoint by phosphorylating CDC25A, resulting in the degradation of CDC25A and subsequent reduction of cyclin-dependent kinase 2 ( CDK2 ) activity in S-phase. Phosphorylation of CDC25C and WEE1 by CHK1 regulates mitosis and the G2/M checkpoint.
In addition, CHK1 also phosphorylates RAD51 on Thr-309, promoting its interaction with BRCA2 during HR.
Elevated CHK1 levels were associated with worse prognosis, disease recurrence, and treatment resistance, further supporting the therapeutic potential of CHK1 inhibition.
WEE1 inhibitors
In response to DNA damage, activated ATR phosphorylates Chk1, which in turn phosphorylates WEE1 and CDC25.
In contrast to CDC25 whose activity is inhibited by phosphorylation, WEE1 is activated and then phosphorylates Tyr15 and Thr14 on downstream CDK1 to inhibit its activity, resulting in G2/M cycle arrest, allowing time for DNA damage repair.
Furthermore, by phosphorylating Tyr15 on CDK1, WEE1 can also prevent the progression from S phase to G2 phase before DNA replication is complete.
Although the rationale for WEE1 inhibitors is clear, their clinical application is limited by their therapeutic window.
Currently, most clinical studies focus on the combined application of WEE1 inhibition with chemotherapeutic drugs.
DNA-PK inhibitors
DNA-dependent protein kinase is a DNA-activated serine/threonine protein kinase that is abundantly expressed in almost all mammalian cells.
DNA-PK is a key protein kinase in DNA repair and is involved in the process of NHEJ.
Upregulation of DNA-PK expression was observed in various tumor types, including gastrointestinal, lung, and hepatocellular carcinoma , and was associated with higher tumor grade and poor prognosis.
The development of DNA-PK inhibitors has mainly focused on the catalytic activity of DNA-PKcs, whereas new anti-DNA-PKcs approaches, such as DNA-PKcs inhibitory microRNAs or inhibitors targeting Ku heterodimers, are based on ATP binding Homology model of the locus.
Currently, most clinical studies of DNA-PK inhibitors focus on their effects in combination with cancer chemotherapy or radiotherapy.
summary
The cellular response to DNA damage is a complex process involving various signaling networks and proteins that are differentially activated or inactivated in specific cancer types.
Increased replication stress and DNA repair defects in tumors present an opportunity for us to treat cancer, making cancer cells more susceptible to DDR inhibition than normal cells.
Currently, drugs targeting the DDR pathway, such as PARP inhibitors, have been clinically applied, and have shown promising prospects in tumor therapy.
Further, determining the optimal dose, combination and schedule of these DDR inhibitors, reducing adverse reactions, and more ideally improving efficacy will be the future direction.
In addition, characterizing each tumor to determine the specific characteristics of its runaway DDR components will aid in the personalized treatment of cancer patients.
references:
1. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm (2020). 2021 Dec; 2(4):654–691.
Tumor therapy targeting DNA repair pathways
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



