Nature: PM2.5 is confirmed to promote lung adenocarcinoma
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Nature: PM2.5 is confirmed to promote lung adenocarcinoma
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Nature: PM2.5 is confirmed to promote lung adenocarcinoma
Air pollution is responsible for 7 million deaths each year.
Particulate matter (PM) is a key component of air pollution.
Small particles (PM2.5) ≤2.5 μm can enter the deep lung and are associated with various adverse health events including lung cancer.
Recently, Nature published an important study, confirming that PM2.5 is significantly related to the occurrence of EGFR mutant lung cancer, and clarifying the pathogenic mechanism of PM2.5.
Lung cancers in nonsmoking individuals (LCINS) and lung cancers in smokers have distinct clinical and molecular features. LCINS is often EGFR-mutant adenocarcinoma, and is more common in women and East Asian patients.
Previously, carcinogens were believed to cause tumors by directly inducing DNA damage. However, the tumor genome characterization of LCINS failed to detect exogenous oncogenic mutation signals.
Therefore, the researchers hypothesized that air pollution may promote inflammatory changes in the microenvironment of lung tissue, resulting in the expansion of mutant clones. This hypothesis is consistent with a two-stage oncogenic model of cancer initiation and progression.
EGFR-driven lung cancer correlates with PM2.5 exposure levels, 3 years of air pollution exposure is sufficient for correlation to emerge
To determine whether air pollution can promote lung cancer without inducing exogenous mutational signatures, studies focused on EGFR-driven lung cancer.
This is due to the high incidence of EGFR-mutant lung cancer in LCINS with low mutational burden.
To explore the relationship between air pollution and EGFR-driven lung cancer incidence, an ecological correlation analysis was performed. Incorporating data from three countries and regions including the UK, South Korea, and Taiwan to explore different PM2.5 air pollution ranges and the incidence of EGFR-driven lung cancer, the results showed that there was a consistent correlation between PM2.5 levels and the incidence of EGFR-driven lung cancer in each region sex.
For each 1 μg m-3 increase in PM2.5 levels, the relative ratio of EGFR-driven lung cancer incidence was 0.63 (P = 0.0028), 0.71 (P = 0.0091) in South Korea, and 1.82 (P = 4.01×10- 6).
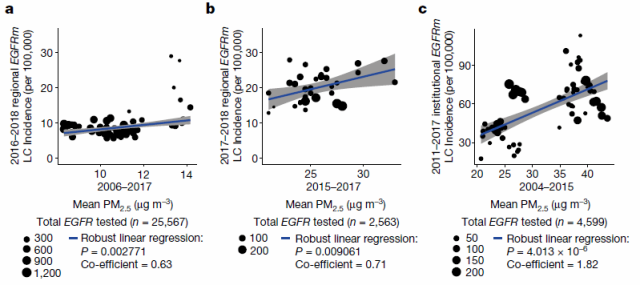
Figure 1 Correlation between lung cancer and air pollution (a) United Kingdom, (b) South Korea, (c) Taiwan
An analysis of 228 female patients with LCINS in British Columbia, Canada, calculated the cumulative exposure to PM2.5 for each individual from birth to current residence, and found that after 3 years of high air pollution exposure, the incidence of EGFR-driven lung cancer cases increased significantly (73% vs 40%, P = 0.03), confirming that 3 years of high PM2.5 exposure is sufficient to increase the incidence of EGFR-driven lung cancer.
Analysis of data from 407,509 participants in the UK Biobank as an independent cohort confirmed that PM2.5 levels were associated with lung cancer incidence (HR 1.08, P≤0.001). In contrast, lung cancer incidence was not associated with outdoor radon levels (HR 0.96, P = 0.262).
Simultaneous interaction tests indicated that smoking and high PM2.5 levels had additive effects on lung cancer risk.
PM mediates lung cancer development through macrophage infiltration
EGFR L858R expression was induced in the lungs of mice (ET mice), after which mice were exposed to physiologically relevant doses of PM or PBS for 3 weeks.
Lung epithelial cells expressing EGFR expanded to form pre-invasive tumors within 10 weeks. The analysis revealed that PM dose-dependently increased the number of pre-invasive tumors.
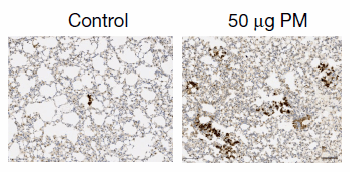
Figure 2. EGFR L858R-positive tumors exposed to PBS and PM
PM also increased the number of pre-invasive tumor cells while EGFR L858R induction was restricted to alveolar type II cells (AT2). Exposure to PM prior to induction of the EGFR L858R mutation also resulted in increased numbers of early tumors, suggesting that PM exposure both before and after gene induction can promote tumorigenesis.
Meanwhile, PM exposure increased the number of adenocarcinomas in the more aggressive doxycycline-induced LUAD EGFR L858R model and also increased the number of hyperplasia in the KRAS G12D LUAD model, suggesting that PM induces tumor Survive.
Exploring the mechanism by which PM promotes EGFR-driven lung carcinogenesis using dynamic spatial analysis of tumor-producing clones in ET mice revealed that expansion of EGFR-mutant cells did not occur during PM exposure, but rather in the post-PM cessation period.
Ten weeks after PM exposure, both the proportion of EGFR L858R cells in the expanded colonies and the number of cells in the colonies increased.
Thus, PM can promote tumorigenesis in 2 ways: by increasing the number of EGFR-mutant cells, or by increasing the proliferation rate of EGFR-mutant cells in early lesions.
The study also found that short-term exposure to PM did not induce DNA mutagenesis.
We also found that the integrity of the immune system is a necessary factor for PM to enhance EGFR-driven lung tumorigenesis, and exposure to PM after EGFR L858R induction in immunodeficient mice did not lead to increased tumors.
The acute myeloid response to PM was observed in immune-intact mice (control) and EGFR mutant mice (ET mice) exposed to PM, and the pan-macrophage marker CD68 was found to increase in the lungs of ET mice, confirming that PM exposure has Higher density of macrophages.
These macrophages were all CD11b+CD68+ interstitial macrophages. The data support the hypothesis of increased lung macrophage infiltration after transient PM exposure.
PM mediates AT2 cell reprogramming
The study further explored the effect of PM exposure on early tumorigenesis and found that the IL-6-JAK-STAT pathway, inflammatory response and graft rejection pathway of EGFR mutant epithelium were only up-regulated after PM exposure.
In ET mice, PM exposure resulted in upregulation of macrophage recruitment genes, including genes encoding IL-1β, GM-CSF, CCL6, and NF-κB and IL-33.
AT2 cells may be the source cells of lung adenocarcinoma, and the bleomycin lung injury model found that keratin 8-positive (KRT8+) subgroup cells are precursor cells that mediate inflammatory signals to drive alveolar regeneration.
These data collectively suggest that PM promotes tumorigenesis in EGFR L858R AT2 cells.
The study also observed upregulation of genes associated with the AT2 progenitor cell state.
The AT2 activated progenitor score increased only in PM-exposed ET mice, suggesting that EGFR L858R AT2 cells were transcriptionally reprogrammed to this progenitor state following PM exposure.
Lung epithelial cells from ET mice exposed to PM were extracted and analyzed for organoid formation.
It was found that EGFR wild-type cells did not show an increase in organoid formation efficiency (OFE) after exposure to PM, while recombinant EGFR L858R cells showed an increase in OFE.
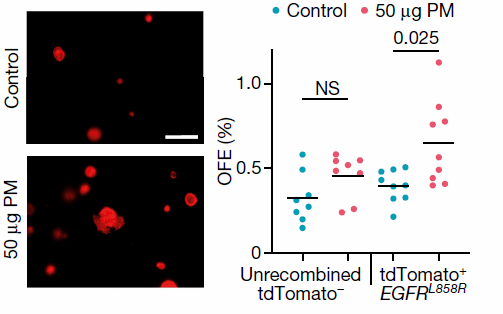
Figure 3 Increased OFE in organoids after exposure to PM
PM-exposed AT2 organoids were KRT8+ and SPC+, consistent with the AT2 progenitor cell state.
The data demonstrate that the combination of in vivo PM exposure and induction of the EGFR L858R driver mutation increases AT2 cell progenitor function, a phenotype not observed in PM-only exposure or EGFR L858R expressing cells.
PM induces macrophages to produce IL-1β, a key signal in EGFR mutant adenocarcinoma
Inflammatory cytokines released by lung macrophages exposed to PM may be important for tumor promotion. AT2 cells were isolated from ET mice not exposed to PM, and the expression of EGFR L858R was induced in vitro, and co-cultured with macrophages exposed to PM or PBS in vivo, and it was found that PM exposed interstitial macrophages and alveolar macrophages increased EGFR mutation OFE in AT2 cells, indicating that the key mediator of PM-induced inflammation comes from macrophages.
Previous reports have shown that IL-1β from lung macrophages is required for the formation of KRT8+AT2 progenitors after bleomycin injury.
Therefore, it is speculated that IL-1β may be an important molecular regulator of tumor development. IL-1β was upregulated in PM-treated lungs, mainly in CD68+ macrophages. Furthermore, IL-1β treatment of EGFR-mutant AT2 cells in vitro resulted in larger KRT8+SPC+ organoids.
To examine the necessity of IL-1β in PM-induced adenocarcinoma formation, the oncogene was expressed in a doxycycline-induced EGFRL858R model and mice were exposed to PM while treated with anti-IL-1β or control antibody. Receiving an anti-IL-1β antibody during PM exposure was sufficient to attenuate EGFR-driven LUAD formation.
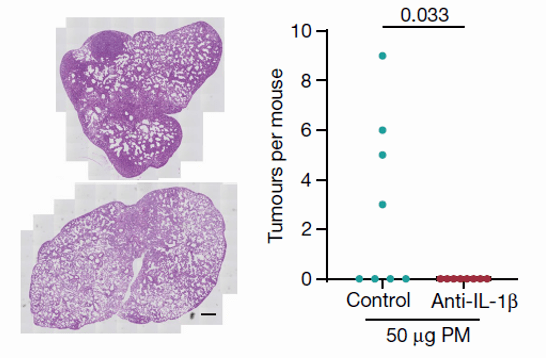
Figure 4 IL-1β antibody can reduce PM exposure-induced EGFR mutant lung cancer
These data establish that PM exposure of macrophages can induce a progenitor-like state in EGFR-mutant AT2 cells, that macrophages are a key source of IL-1β following PM responses, and that IL-1β signaling is required to facilitate PM-mediated EGFR-driven LUAD .
Oncogene mutations also present in healthy lungs
The study selected non-cancerous lung tissue from lung cancer patients for detection, and found that oncogene driver mutations such as EGFR can exist in normal lung tissue.
Mutations such as EGFR or KRAS were not associated with smoking status, whereas age was significantly associated with mutational values .
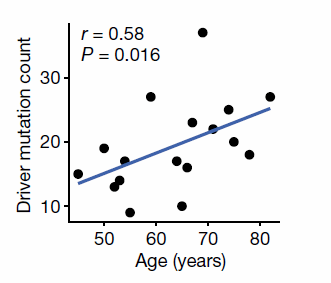
Figure 5 The number of driver mutations increases with age
Sum up
- The study found a correlation between the incidence of EGFR-mutant lung cancer and increased PM2.5 levels. Time analysis showed that 3 years of PM2.5 exposure was sufficient to increase the risk of EGFR-driven lung cancer.
- PM induces the transformation of EGFR-mutant AT2 cells into a progenitor state through IL-1β released from macrophages, promoting the development of lung cancer, and antibodies targeting IL-1β can reduce the incidence of lung cancer.
- Studies have confirmed that the key contributor to cancer development is not just the acquisition of genetic mutations, but also the balance of endogenous and exogenous mechanisms. There is a clear link between air pollution and lung cancer, and the research informs public health policy to limit particulate matter releases in cities.
Summary
The study found a correlation between the incidence of EGFR-mutant lung cancer and increased PM2.5 levels.
Time analysis showed that 3 years of PM2.5 exposure was sufficient to increase the risk of EGFR-driven lung cancer.
PM induces the transformation of EGFR-mutant AT2 cells into a progenitor state through IL-1β released from macrophages, promoting the development of lung cancer, and antibodies targeting IL-1β can reduce the incidence of lung cancer.
Studies have confirmed that the key contributor to cancer development is not just the acquisition of genetic mutations, but also the balance of endogenous and exogenous mechanisms.
There is a clear link between air pollution and lung cancer, and the research informs public health policy to limit particulate matter releases in cities.
references
Hill W, et al. Lung adenocarcinoma promotion by air pollutants. Nature. 2023;616(7955):159-167
Nature: PM2.5 is confirmed to promote lung adenocarcinoma
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



