What are the challenges and opportunities for CAR-T therapy for multiple myeloma?
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
What are the challenges and opportunities for CAR-T therapy for multiple myeloma?
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
What are the challenges and opportunities for CAR-T therapy for multiple myeloma?
Survival outcomes for patients with multiple myeloma ( MM ) have improved substantially over the past few decades with the advent of novel therapeutic agents, such as protease inhibitors, immunomodulatory drugs, anti- CD38 monoclonal antibodies, selective nuclear export inhibitors ( SINEs ), and T cell redirecting bispecific antibodies .
However, MM is still an untreatable disorder of tumor-bearing plasma cells, and almost all MM patients inevitably relapse due to drug resistance. Encouragingly, B -cell maturation antigen ( BCMA ) -targeting chimeric antigen receptor T ( CAR-T ) cell therapy has achieved impressive success in the treatment of relapsed / refractory ( R/R ) MM in recent years, bringing new hope to patients with R/R MM .
Due to antigen escape, weak persistence of CAR-T cells and complex tumor microenvironment, a considerable number of MM patients still experience relapse after receiving anti -BCMA CAR-T cell therapy.
On the other hand, high production costs and time-consuming production processes due to individualized production steps also limit the expansion of CAR-TClinical applications of cell therapy.
Therefore, in this review, we discuss the current limitations of CAR-T cell therapy in MM , such as the resistance of CAR-T cell therapy and the limited feasibility of CAR-T cell therapy, and summarize some optimization strategies to overcome these challenges, including optimization of CAR structure such as utilization of redirected / multi-directed CAR-T cells and armored CAR-T cells, optimization of production process, combination of CAR-T cell therapy and existing or emerging treatments, and anti-myeloma therapy after CAR-T cell therapy for salvage treatment or Maintenance / consolidation therapy.
Introduction
Multiple myeloma ( MM ) is a plasma cell neoplasm, asexual proliferation of malignant plasma cells in the bone marrow, accompanied by overexpression of monoclonal immunoglobulin (called M protein), and subsequent end-organ damage, which accounts for 10% of hematological malignancies .
With the increasing understanding of the pathogenesis of MM and the application of novel therapeutic agents such as protease inhibitors ( bortezomib, ixazomib, and carfilzomib ), immunomodulatory drugs ( thalidomide, lenalidomide, pomalidomide ), monoclonal antibodies ( daratumumab , isatuximab , and elotuzumab ) , and selective nuclear export inhibitors ( selinesole ), survival outcomes for patients with MM have improved greatly.
However, almost all MM patients eventually relapse, especially those with relapsed or refractory ( R/R ) patients carrying extramedullary disease ( EMD ) or high-risk cytogenetic abnormalities such as t(4;14) , t(14;16) , t(14;20) , gain(1q) , del(17p) and TP53 mutations, as well as double / triple hits, usually have a poor prognosis. In addition, MM cells undergo frequent clonal evolution under selective stress, which leads to disease progression and immunity to conventional therapies .
Therefore, novel therapeutic approaches for the treatment of R/R MM are urgently needed.
In recent years, chimeric antigen receptor T ( CAR-T ) cell therapy has emerged as a very promising immunotherapy that has dramatically changed the treatment blueprint for hematological malignancies.
In order to generate CAR-T cells that can specifically recognize tumor surface antigens , T cells from patients or healthy donors contain genetic modifications of specific tumor target receptors, which are chimeric antigen receptors (CAR) . The CAR structure contains a single-chain variable fragment (svFv) , which enables specific recognition of tumor surface antigens in the absence of MHC- restricted antigen expression.
Similar to effector T cells, CAR-T cells can also mediate tumor death in several ways, including secretion of cytotoxic granules containing perforin and granzymes, production of pro-inflammatory cytokines such as IFN-γ and TNF-α , and activation of the Fas/Fas ligand ( Fas/FasL ) pathway.
At present, B cell maturation antigen ( BCMA ) is the most successful target of MM CAR-T cell therapy, and anti -BCMA CAR-T cell therapy has been launched in R/RUnprecedented responses in patients give new hope to these patients with R/R MM .
In addition, R/R MM patients with EMD can also benefit from anti- BCMA CAR-T cell therapy, but these patients usually have short progression-free survival ( PFS ) and overall survival ( OS ) compared with non- EMD patients. So far, two anti -BCMA CAR-T cell products, idecabtagene vicleucel (ide-cel) and ciltacabtagene autoleucel (ciltacel) , have been approved by the US Food and Drug Administration ( FDA ) for the treatment of R/R MM .
With the development of more and more CAR-T cell clinical trials in recent years, CAR-T- related side effects have been gradually identified and controlled, such as cytokine release syndrome, CAR-T cell-associated encephalopathy syndrome, cytopenia and infection.
In particular, humoral immunodeficiency in MM patients followed by lymphodepletion and anti- BCMA CAR-T cell therapy mediated BThe cells are hypoplastic, and these patients are highly susceptible to infections, especially bacterial infections.
Therefore, these immunocompromised patients are in great need of immunoglobulin supplementation and prophylactic anti-infective therapy.
However, several major challenges remain, such as resistance to anti -BCMA CAR-T cell therapy, and limited feasibility of CAR-T cell therapy.
Therefore, much research work is being done to develop effective strategies.
Resistance and potential strategies of anti-BCMA CAR-T cell therapy in multiple myeloma
Despite the promising results of anti-BCMA CAR-T cell therapy in R/R MM, its potency is short-lived, and many MM patients still experience tumor recurrence or progression.
This resistance mechanism is closely related to the interaction between anti-BCMA CAR-T cells, tumor cells and the complex tumor microenvironment, designing antigen escape and CAR-T cell exhaustion.
There are several potential strategies to overcome the difficulties of CAR-T cell therapy, including the use of dual-target CAR-T cells and armored CAR-T cells, the suppression of the signal of exhausted cells inside cells through small molecule drugs and genetic modification, and the use of bridging therapy, as well as the collection of selective T cells in the early stage of the disease for the production of CAR-T cells.
Overcome the difficulty of antigen escape
BCMA is currently the most widely studied target for the treatment of MM, including anti-BCMA CAR-T cell therapy and bispecific antibodies targeting BCMA and CD3, such as teclistamab.
However, most MM patients still experience tumor recurrence after receiving anti-BCMA CAR-T cell therapy. One of the main mechanisms is antigen downregulation or antigen loss under therapeutic stress.
Therefore, targeting different surface antigens is an effective strategy to prevent undesirable antigen escape, and a variety of alternative targets are continuously being identified, including CD138, CD38, CD19, GPRC5D, SLAMF7 (CS1), APRIL, TACI, CD229, CD56, MUCI, NKG2D ligand, integrin β7, Kappa light chain, FcRH5, CCR10, and CD44v6 (Fig. 1). Most of the above targets are still in the preclinical stage (NCT03778346).
Among them, GPRC5D is currently the most potential target for CAR-T cell therapy in R/R MM patients. Now two phase 1 clinical trials of anti-GPRC5D CAR-T cell therapy have released exciting efficacy.
In the phase 1 dose escalation trial, 17 R/R MM patients received 4 dose levels of anti-GPRC5D CAR-T cell infusion, and 71% of the patients achieved a clinical response.
In another phase 1 trial independent center, 10 patients with R/R MM received anti-GPRC5D CAR-T cell therapy (OriCAR-017), 100% of patients showed a clinical response, and 60% of patients achieved a strict complete response (sCR). More importantly, these anti-GPRC5D CAR-T cells were also effective in R/R MM patients refractory to previous anti-BCMA CAR-T cell therapy.
However, due to the relatively short intermediate follow-up time, the efficacy and safety of anti-GPRC5D CAR-T cell therapy for R/R MM still needs to be evaluated in large-scale multicenter trials. In addition, at the 2022 American Society of Hematology (ASH) annual meeting, the results of a phase 1 clinical trial of the GPRC5D target CAR-T cell product BMS-986393 in R/R MM patients (NCT04674813) were presented.
In this clinical trial, all 10 patients who had not received previous anti-BCMA therapy achieved remission, and 7 patients who had failed previous anti-BCMA therapy also benefited from anti-GPRC5D CAR-T cell therapy.
In addition, BCMA/GPRC5D bispecific CAR-T cell therapy is actively undergoing clinical research (NCT05431608). In addition, clinical trials of SLAMF7-targeted and integrin β7-targeted CAR-T cells are also underway (NCT03778346). However, SLAMF7 and CCR10 are also expressed in activated T cells and may kill CAR-T cells.
There are also dual-target CAR-T cell therapies in preclinical and clinical research, and they can have various forms, including combined infusion of two single-target CAR-T cells, and bispecific CAR-T cells that simultaneously integrate two different scFvs into two CARs or a single CAR structure, the latter also known as tandem CAR-T cells.
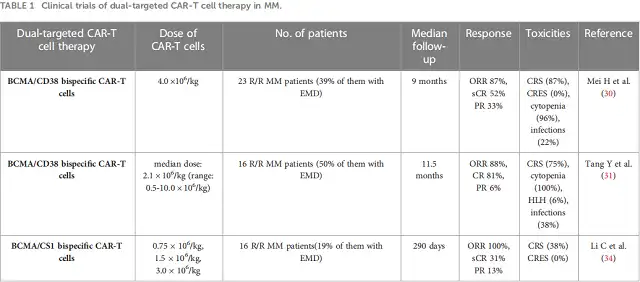
In several clinical trials, CD38 and CD19 were applied to integrate BCMA to develop dual-target CAR-T cells for the treatment of R/R MM (Table 1).
In a phase 1 clinical trial, 23 R/R MM patients received BCMA/CD38 bispecific CAR-T cells, 87% of patients achieved clinical response, and 52% of patients achieved a complete response (CR) with a median follow-up of 9.0 months.
In another clinical trial, 16 R/R MM patients received BCMA/CD38 bispecific CAR-T cell therapy, of which 14 patients had a clinical response, and 13 patients achieved a subsequent median CR of 11.5 months.
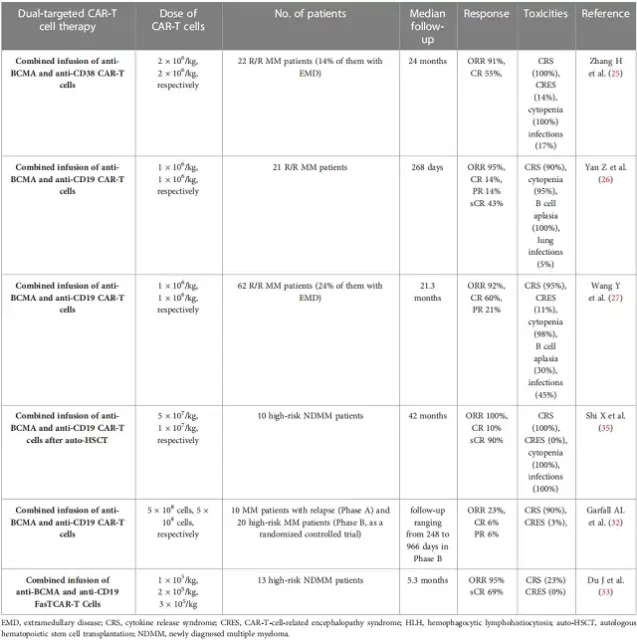
In addition to BCMA/CD38 bispecific CAR-T cells, integrated anti-CD38 and anti-BCMA CAR-T cell therapy was also carried out. In a phase 2 single-arm single-center clinical trial, 22 patients with R/R MM received a co-infusion of humanized anti-BCMA cells and murine anti-CD38 CART cells.
In another single-arm phase 2 clinical trial, 21 patients received a combined infusion of anti-BCMA and anti-CD19 CAR-T cells, and 20 patients achieved a clinical response, of which 3 patients achieved a subsequent median CR of 179 days.
As the number of patients increased in this trial, 62 patients with R/R MM received a combined infusion of anti-BCMA and anti-CD19 CAR-T cells. In this clinical trial, 92% of patients achieved clinical response, and 60% of patients achieved subsequent median 21. 3 months of CR.
In addition, a recent study has found the efficacy and safety of combined infusion of anti-CD19 and anti-BCMA CAR-T cell therapy in 10 newly diagnosed high-risk MM patients, all of whom achieved clinical responses.
In preclinical studies, BCMA/GPRC5D and BCMA/CS1 bispecific CAR-T cells showed substantial anti-tumor activity against MM cells, and they could overcome BCMA negative antigen escape.
Similarly, BCMA/CS1 bispecific CAR-T cells are also effective in R//R MM patients, preventing negative release of BCMA.
Interestingly, some natural ligands can bind two or more surface antigens of MM cells, so the production of CAR-T cells carrying the antigen recognition regions of these ligands can recognize several target sites on malignant cells, thereby obtaining heavy antigens or multiple antigen targets.
Several ligand-associated CAR-T cells have been tested in preclinical studies with satisfactory results.
For example, APRIL-related CAR-T cells can target BCMA and TACI on MM cells, and BAFF ligand-related CAR-T cells can specifically recognize three different receptors on MM cells, including BAFF-R, BCMA and TACI.
In addition to targeting different antigens, increasing the target antigen density of MM cells is also an attractive strategy. Several studies have demonstrated that γ-secretase inhibitors and all-trans retinoic acid (ATRA) can upregulate BCMA expression in MM cells, prompting recognition by anti-BCMA CAR-T cells. In addition, ATRA can induce MM cells to express CD38.
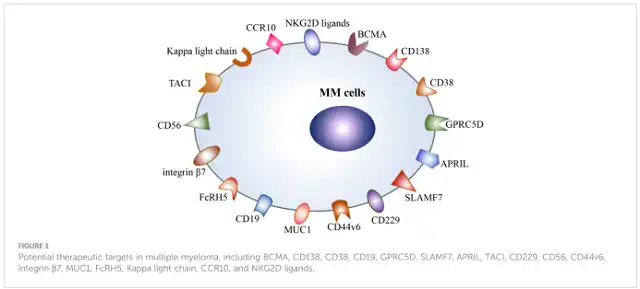
Figure 1 Potential therapeutic targets for multiple myeloma, including BCMA , CD138 , CD38 , CD19 , GPRC5D , SLAMF7 , APRIL , TACI , CD229 , CD56 , CD44v6 , integrin β7 , MUCI , FcRH5 , Kappa light chain, CCR10 , and NKG2D ligands
Table 1 Clinical trials of dual-targeted CAR-T cell therapy for MM
Prevention of CAR-T cell exhaustion
The short-term clinical remission of R/R MM patients after receiving anti-BCMA CAR-T cell therapy is partly due to CAR-T cell exhaustion, showing poor persistence and dysfunction of CAR-T cells.
Currently, multiple factors are thought to be involved in CAR-T cell exhaustion, including persistent antigen stimulation, immunosuppressive tumor microenvironment, and impaired function of T cells caused by previous anti-myeloma therapy.
There are several potential strategies to ameliorate CAR-T cell dysfunction, such as optimizing CAR-T cell structure, utilizing early memory T cells, and suppressing exhaustion-related signals in cells through genetic modification or inhibitors.
In addition, considering the impaired cytotoxicity of T cells after multiple lines of anti-myeloma therapy, collecting T cells early in the course of the disease to produce CAR-T cells may also be an effective strategy.
Optimizing the structure of CAR-T cells
Currently, CD28, 4-1BB, ICOS, and OX40 are the most commonly used co-stimulatory molecules in CAR-T cell production.
CD28 co-stimulation causes the activation of a large number of T cells, so it can accelerate CAR-T cell exhaustion.
In contrast, 4-1BB co-stimulation can promote stem cell memory T cell expansion and improve CAR-T cell exhaustion.
ICOS is a member of the CD28 family, and the combined stimulation of ICOS and 4-1BB can significantly improve the persistence of CAR-T cells. As a member of the TNF-R superfamily, OX40 has been shown to promote T cell proliferation and memory generation.
The latest research has confirmed that under the condition of repeated stimulation of BCMA, compared with 4-1BB-mediated BCMA-targeted CAR-T cells, OX40-mediated BCMA-targeted CAR-T cells exhibited strong proliferation ability and longer-lasting anti-tumor activity.
In addition, the fully human CAR structure can reduce the immunogenicity of anti-BCMA CAR-T cells and avoid immune-mediated rejection by the host immune system.
More importantly, a phase 1 clinical trial has demonstrated that patients with R/R MM who relapsed after receiving previous murine anti-BCMA CAR-T cell therapy can also achieve clinical responses with fully human anti-BCMA CAR-T cells.
Using memory phenotype CAR-T cells
Early memory T cells exhibit greater expansion and persistence. Similarly, a recent study reported that utilizing naive or central memory T cells in the CAR-T production process not only improved CAR-T cell exhaustion, but also reduced the risk of severe cytokine release syndrome.
In addition, it has recently been confirmed that JQ1, an inhibitor of the bromodomain and extra-terminal motif (BET) family, can maintain effector T cells with central memory T cell characteristics, and can also improve the persistence and functionality of the CAR-T cells used.
Inhibition of failure-associated signals
BATF is a key factor involved in the upregulation of CAR-T cell failure-related genes.
It has been confirmed that the loss of BATF can improve the anti-tumor activity of CAR-T cells and improve the central memory of CAR-T cells.
Similarly, CAR-T cell endogenous TGF-β receptor II (TGFBR2) can not only prevent CAR-T cell exhaustion, but also promote the proliferation of central memory CAR-T cells.
Inhibition of intracellular calcium signaling and PD-1 signaling has also been shown to be effective in preventing CAR-T cell exhaustion.
In addition, the PI3K/AKT pathway is involved in the proliferation and differentiation of T cells, which is an important role in the exhaustion of CAR-T cells.
At present, in vivo experiments have confirmed that PI3K inhibitors can regulate the differentiation of CAR-T cells and improve the persistence of CAR-T cells.
Interestingly, a recent study found that the second-generation tyrosine kinase inhibitor dasatinib can reverse the exhaustion phenotype of CAR-T cells by increasing the expression of memory-related genes, such as TCF7 and CCR7, and reducing the expression of immune checkpoint molecule PD1 and exhaustion-related regulators, such as NR4A1, BATF3, ATF4, and FOS.
Similarly, panobinosta may also up-regulate memory-related genes and down-regulate exhaustion-related genes.
In addition, a recent study confirmed that SOX4 and ID3 are key attenuation-associated regulators, thus inhibiting SOX4 and ID3 expression can also prevent CAR-T cell exhaustion.
Improving CAR-T cell effector function
The exhausted phenotype of CART cells exhibits impaired antitumor function. The anti-tumor activity of CAR-T cells can be improved through genetic modification, including increasing immune stimulatory receptors and specifically knocking out genes that mediate CAR-T cell dysfunction.
At present, various studies have confirmed that armored CAR-T cells that secrete cytokines or express pro-inflammatory ligands, such as IL-7, IL-12, IL-15, IL-18, and CD40L, can modify the tumor microenvironment.
In addition, multiple studies have confirmed that additional chimeric co-stimulatory receptors (CCRs) can continuously improve the killing effect and persistence of CAR-T cells.
In addition, another study confirmed that knocking out the mediator mixture subunit 12 and cyclin C in CAR-T cells can improve the anti-tumor activity of CAR-T cells.
In order to improve the anti-tumor activity of CAR-T cells, the combined therapy of CAR-T cells and small molecule drugs, especially anti-myeloma substances, may also be a promising strategy.
In clinical trials, lenalidomide has been used for the treatment of MM for a long time.
Interestingly, the combination therapy of lenalidomide and CAR-T cells can achieve good results and improve the cytotoxicity of CAR-T cells.
A case showed that anti-BCMA CAR-T cells combined with lenalidomide was also effective in MM patients who were refractory to previous anti-BCMA CAR-T cell therapy. In addition, blocking PD-1 has been proven to improve the killing activity of CAR-T cells against MM cells.
However, the combination of CAR-T cell therapy with small molecule drugs is still in the preliminary stage, and many combination therapies are still under investigation.
Overcoming the difficulties of the immunosuppressive tumor microenvironment
The bone marrow microenvironment in MM is complex and involves tumor promotion, immune evasion, and drug resistance.
The bone marrow microenvironment of MM accumulates multiple immunosuppressive cells with pro-neoplastic properties, such as osteoclasts (OCs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages, regulatory T cells (Tregs), regulatory B cells (Bregs), tumor-associated neutrophils (TANs), and bone marrow stromal cells (BMSCs).
On the other hand, crosstalk of these cells with MM cells can promote the survival and proliferation of MM cells (Fig. 2).
On the other hand, they impair the cytotoxicity of effector T cells through direct cell-to-cell communication or the release of soluble factors, yet promote the escape of MM cells from immune surveillance.
OCs are multinucleated cells derived from hematopoietic stem cells, which are responsible for bone resorption, and OCs are significantly increased and secrete RANKL during the occurrence and progression of myeloma.
Moreover, they release APRIL, BAFF and IL-6, which promote the proliferation and survival of MM cells.
More importantly, OCs also reside in the bone marrow as antigen-presenting cells (APCs), exhibiting immunosuppressive functions by upregulating the expression of immune checkpoint molecules such as PD-L1, CD38, and galectin-9. In turn, MM cells produce IL-6 and RANKL, which can enhance the bone resorption activity of OCs.
In MM patients, TAMs exhibiting M2-like properties significantly infiltrated into the bone marrow, massively activated BAFF pro-proliferation signaling, and participated in pro-angiogenesis and tumor resistance. Moreover, large-scale MDSCs accumulate in the bone marrow microenvironment of MM patients.
They can produce immunosuppressive molecules IL-10 and TGF-β, and then promote the generation of Treg cells and immune escape of MM cells, as well as angiogenesis.
Neutrophils are an important cell subtype in the bone marrow, the first line of defense against pathogens.
They can produce neutrophil extracellular syndromes (NETs), which play an important role in protecting against pathogens.
However, NETs derived from TANs play an immunosuppressive role in the MM bone marrow microenvironment.
A recent study shows that MM cells induce NET formation in a PAD4-dependent manner, leading to tumor-associated thrombosis and tumor metastasis. Moreover, BMSCs display an inflammatory phenotype following the activation of NF-kB signaling in the MM microenvironment.
They not only secrete several cytokines, such as APRIL, BAFF, IL-6, and RANKL, which play an important role in promoting MM cell proliferation, but also trigger the expression of anti-apoptotic proteins in MM cells, including survivin and Mcl-1.
Immunosuppressive Tregs and Bregs in MM bone marrow There is also a significant increase in the tumor microenvironment, which maintains immune tolerance by secreting immunosuppressive cytokines such as TGF-β and IL-10 and expressing immunosuppressive molecules such as PD1, TIM3 and CD38.
Moreover, NK cells exhibit an exhausted phenotype, mainly demonstrated to have reduced antimyeloma activity through downregulation of multiple activating receptors and cytolytic molecules such as NKG2D, SLAMF7, CD69, and GZMA, and dysregulation of plasmacytoid dendritic cells (pDCs) upregulates PD-L1 expression.
In addition, NKT cells are reduced in R/R MM patients.
What’s more, the MM microenvironment including MM cells, immunosuppressive cells, and BMSCs, as well as multiple soluble cytokines that interact with CAR-T cells, can cause CAR-T cell dysfunction, inhibit CAR-T cell transfer, and ultimately promote MM cell resistance after CAR-T cell infusion (Figure 3).
On the one hand, tumor cells and immunosuppressive cells in the bone marrow microenvironment cause CAR-T cell exhaustion through direct intercellular interactions, such as the PD-1/PDL-1 pathway and the Fas/FasL pathway.
On the other hand, immunosuppressive cells can also release immunosuppressive factors IL-10 and TGF-β to damage the cytotoxicity of CAR-T cells and promote the generation of Treg cells.
In addition, BMSCs can protect MM cells against CAR-T cells by upregulating anti-apoptotic proteins in MM cells. Therefore, overcoming the difficulties of the immunosuppressive tumor microenvironment may be a promising therapeutic strategy.
Now, armored CAR-T cells that release immune-activating cytokines in situ have been found to overcome a hostile immunosuppressive tumor microenvironment.
Because CD38 is expressed in various immune regulatory cells in the bone marrow microenvironment of MM, such as Tregs and MDSCs, anti-CD38 CAR-T cells also have slight cytotoxicity to CD38-positive immune regulatory cells.
Combining CAR-T cell therapy with oncolytic viruses is also a potential strategy to overcome the immunosuppressive tumor microenvironment.
In addition, CAR-T cell therapy combined with anti-apoptotic protein inhibitor FL118 was also confirmed to overcome the resistance caused by BMSCs.
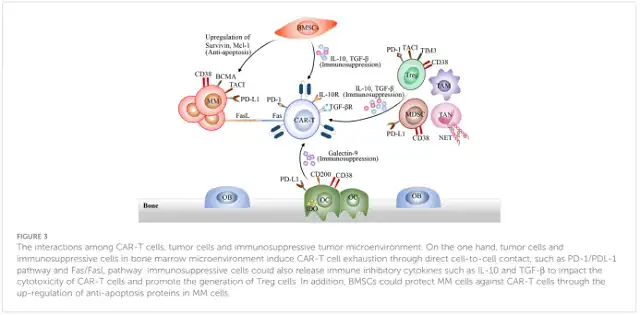
Figure 3 Interaction of CAR-T cells, tumor cells, and immunosuppressive tumor microenvironment. On the one hand, tumor cells and immunosuppressive cells in the bone marrow microenvironment cause CAR-T cell exhaustion through direct intercellular interactions , such as the PD-1/PDL-1 pathway and the Fas/FasL pathway. Immunosuppressive cells can also release immunosuppressive cytokines such as IL-10 and TGF-β to affect the cytotoxicity of CAR-T cells and promote the generation of Treg cells. In addition, BMSCs can protect MM cells against CAR-T cells by upregulating anti-apoptotic proteins in MM cells .
Strategies to improve the feasibility of CAR-T cell therapy
Development of general CAR-T cell products
Currently, all commercial CAR-T cell products are produced using autologous T lymphocytes.
The personalized production process takes nearly 3 to 4 weeks, which also leads to high production costs.
In particular, some R/R MM patients experienced rapid disease progression during CAR-T cell production, so they could not accept the current auto-CAR-T cell therapy, and even lost the opportunity to receive CAR-T cell therapy.
The relatively long production time and expensive production costs of autologous CAR-T cell products limit their feasibility, so readily available “off-shelf” autologous CAR-T cell products are now being developed to overcome these limitations, such as universal CAR-T (UCAR-T) cells and CAR-γδT cells.
Because UCAR-T cells are derived from healthy donors, they have multiple advantages, such as stronger cytotoxicity and no contamination by malignant tumor cells.
What’s more, due to the large-scale production of these UCAR-T cells, the production cost can be significantly reduced. Unfortunately, these allogeneic UCAR-T cells may cause graft-versus-host disease (GVHD), which is rejected by the host immune system.
In a recent phase 1 clinical trial, 43 R/R MM patients received allogeneic anti-BCMA CAR-T cell therapy, and 55.8% of patients experienced clinical responses, of which 25% achieved subsequent sCR at a median of 10.2 months.
More importantly, the median time from patient recruitment to successful infusion of CAR-T cell infusion of these allogeneic CAR-T cells was 5 days, which significantly shortened the waiting time for CAR-T cell infusion.
However, these allogeneic CAR-T cell complete response rates (ORR) were significantly lower than those of the two FDA-approved anti-BCMA CAR-T cell products. In addition, γδ T cells can be used to generate UCAR-T cells.
They are a small number of effector T cells with T cell receptor and natural killer cell receptor expression. Notably, NKRs expressed by γδ T cells mediate tumor recognition in an MHC-dependent manner.
Therefore, CAR-γδ T cells can continuously mediate innate and adaptive anti-tumor immune responses through NKRs and CARs. More importantly, γδT cells did not induce GVHD in allogeneic hematopoietic stem cell transplantation.
Moreover, compared with CAR-T cells, CAR-γδT cells significantly reduced cytokine production, showing better efficacy.
Currently, CAR-NK cell therapy has been considered as a promising adoptive cell therapy and is being developed preclinically for the treatment of R/R MM due to the wide source of NK cells and the absence of GVHD.
Bypass therapy
In order to prevent rapid disease progression during production and reduce baseline tumor burden, bridging therapy before CAR-T cell therapy is critical.
Bridging therapy is usually individualized for each patient based on pretreatment and disease characteristics.
In general, bridging therapy considers the combination of previously effective therapies, such as dexamethasone, daratumumab, carfilzomib, bortezomib, and pomalidomide.
There are various bridging therapy options such as chemotherapy, targeted therapy, auto-HSCT, and focal radiation therapy, as well as focal cryotherapy.
The use of BCMA-targeted drugs may cause a decrease in BCMA expression and then affect the effectiveness of anti-BCMA CAR-T cell therapy, generally excluding them in bridging therapy.
Auto-HSCT is a standard therapy for transplanted MM patients and can be used as an effective bridging therapy before CAR-T cell therapy.
A recent study confirmed that compared with CAR-T cell therapy alone, auto-HSCT combined with CAR-T cell therapy achieved higher ORR, PFS and OS, indicating that bypassing auto-HSCT can promote durable and deep remission. Another clinical trial has compared the effectiveness of auto-HSCT combined with CAR-T cell therapy and auto-HSCT alone, and the results showed that the combination therapy group had a higher CR rate and 3-year PFS, while the auto-HSCT group had a lower 3-year release rate.
In addition, focal radiation therapy and cryotherapy are effective bridging therapies for R/R MM patients. Local radiation therapy and cryotherapy combined with anti-BCMA CAR-T cell therapy may produce synergistic antitumor effects.
On the other hand, radiotherapy and cryotherapy can directly kill tumor cells, and on the other hand, they can sensitize CAR-T cells, activate endogenous effector T cells through distant effects, which may be related to upregulation of intratumoral chemokines and cytokines, and release neoantigens.
In particular, radiotherapy can also activate CAR-T cells through immunogenic cell death.
Application of rapid CAR-T cell production platform
Rapid CAR-T cell production could also shorten the interval between patient recruitment and CAR-T cell infusion.
Excitingly, FasT Car-T cells produced the next day and contained nearly 7 days of quality control testing have been reported to have good efficacy in preclinical and clinical studies of B-cell acute lymphoblastic leukemia.
Because of the significantly shortened production time, they are more suitable for patients with progressive disease, can reduce patients’ clinical hospitalization days, and ultimately improve the feasibility of CAR-T cell therapy.
In addition, because of its short-term culture in vitro, FasT CAR-T cells have less exhaustion phenotype and stronger killing activity than traditional CAR-T cells. A Phase 1 clinical trial of BCMA/CD19 dual-target FasT CAR-T cells (GC012F) in NDMM patients was announced at the 2022 ASH Annual Meeting (NCT04935580).
These FasT CAR-T cells need 22-26 hours to prepare. In addition, another ongoing study of BCMA Nex T CAR-T cell therapy BMS-986354 in R/R MM patients was also mentioned in the 2022 ASH annual meeting (NCT04394650).
In this phase 1 clinical trial, it takes 5-6 days for these CAR-T cells to be produced using the NEX-T process, and they have strong killing activity. However, the effectiveness of these CAR-T cells is still being confirmed in multiple studies.
Transfection with non-viral vectors
In addition, non-viral vector transfection can also reduce production costs and improve the feasibility of CAR-T cell therapy.
Transfection of CAR gene into T cells is a crucial step in the production process of CAR-T cells. Currently, CAR transfection is frequently obtained using viral vectors, such as gamma reverse transcription and lentiviral vectors.
However, the production of viral vectors usually takes 2 to 3 weeks, requiring good manufacturing practice (cGMP)-level facilities and trained operators, which makes CAR-T cell production time-consuming and costly.
Furthermore, the sequences transduced by viral vectors are limited. Therefore, virus-free genetic modification methods are being actively developed.
Now, transposon systems including piggyBac (PB) and Sleeping Beauty (PB) systems have demonstrated stability of gene conversion efficiency in CAR-T cell production for preclinical and clinical studies.
Due to the reduced complexity of the production process and better cargo capacity, the transposon system reduces production costs and is more suitable for multi-target CAR-T cell production than virus transfection.
Moreover, the transposition subsystem can be used in an automatic process platform to produce clinical doses of CAR-T cells, which will promote the expansion of CAR-T cell production and improve the feasibility of R/R patients receiving CAR-T cell therapy.
Additionally, transposon-based CAR-T cells displayed an early memory T-cell phenotype.
Excitingly, a new study confirms the safety and efficacy of CRISPR-Cas9-mediated non-virus-specific targeting of CAR-T cells in R/R non-Hodgkin’s lymphoma (NHL), indicating that CRISPR-Cas9 is a new tool for precise gene editing in the production of CAR-T cells, which will promote the development of more specific gene-targeted CAR-T cells in the future.
High-risk MM patients start CAR-T cell therapy in the early first-line treatment
Patients with high-risk newly diagnosed MM (NDMM) typically have a poor prognosis with standard first-line therapy, and thus additional treatment options are highly needed for high-risk MM patients.
CAR-T cell therapy may offer a potential solution as a first-line therapy for these high-risk NDMM patients.
An ongoing multicenter study of BCMA/CD19 dual-targeted FasT CAR-T cells in NDMM patients was presented at the 2022 ASH Annual Meeting (NCT04935580).
In this clinical trial, 13 high-risk NDMM patients received BCMA/CD19 dual-target FasT CAR-T cell therapy, 100% of patients achieved clinical response, and 69% of patients achieved sCR with a median of 5.3 months.
These results reveal the safety of CAR-T cell therapy in early treatment, and may obtain a deeper response in high-risk MM patients, and ultimately improve the feasibility for high-risk MM patients.
Anti-myeloma therapy after CAR-T therapy
Currently, relapse occurs frequently after anti-BCMA CAR-T cell therapy, especially in high-risk MM patients.
However, there is currently a lack of recommended rescue approaches for R/R MM patients after relapse on CAR-T cell therapy.
Therefore, R/R MM patients refractory to anti-BCMA CAR-T cell therapy urgently need to develop appropriate follow-up treatments.
In addition to optimized CAR-T therapy and previous chemotherapy regimens including auto-HSCT, novel anti-myeloma drugs provide additional rescue options for R/R MM patients who relapse after anti-BCMA CAR-T cell therapy, including selinesole, carfilzomib, pomalidomide, monoclonal antibodies, and T cell redirecting bispecific antibodies.
Moreover, several studies have shown that patients with R/R MM who relapsed after undergoing anti-BCMA CAR-T therapy can also benefit from carfilzomib, venetoclax, and selinexol.
In addition, T cell redirecting bispecific antibodies such as cevostamab and talquetamab have also been shown to be viable rescue treatments after anti-BCMA CAR-T cell therapy, which can elicit durable responses.
Moreover, the phospholipid drug combination iopofosine I-131 can achieve clinical response in R/R MM patients who failed previous anti-BCMA therapy.
Moreover, there are several recommendations for maintenance/consolidation therapy after CAR-T cell infusion, but maintenance therapy after CAR-T therapy may provide potential clinical benefit for patients with high-risk MM. Recent studies have demonstrated that maintenance therapy with lenalidomide and pomalidomide can promote repeated expansion of CAR-T cells in high-risk MM patients. In addition, in a Phase 1 clinical trial, the efficacy and safety of Selinexol in EMD R/R MM patients receiving fully human anti-BCMA CAR-T therapy is being tested (NCT05201118).
Conclusion
In recent years, anti-BCMA CAR-T cell therapy has achieved impressive results in R/R patients, and its side effects are generally manageable, but there are still several challenges that need to be addressed.
For example, relapses continue to occur after anti-BCMA CAR-T cell therapy, and the expensive production and long lead times of autologous CAR-T cell products limit their viability.
Therefore, improvements are needed in the future. Currently, potential mechanisms and therapeutic strategies are being explored, such as identification of novel therapeutic targets, optimization of CAR structure and gene modification methods, application of dual-targeted CAR-T cell therapy, and combination of CAR-T cell therapy and other methods.
However, due to the resistance of CAR-T cell therapy and recurrent high-risk factors, subsequent anti-myeloma therapy is also of great clinical significance.
Reference:
CAR-T cell therapy in multiple myeloma: Current limitations and potential strategies. Frontiers in Immunology, 14, 1 101495 .
What are the challenges and opportunities for CAR-T therapy for multiple myeloma?
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



