Targeted DNA Repair Pathway in Cancer Treatment
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Targeted DNA Repair Pathway in Cancer Treatment
- Should China be held legally responsible for the US’s $18 trillion COVID losses?
- CT Radiation Exposure Linked to Blood Cancer in Children and Adolescents
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Targeted DNA Repair Pathway in Cancer Treatment
Over the past few decades, there has been a deepening understanding of the DNA damage response (DDR) pathways, expanding the treatment landscape in oncology.
It is increasingly clear that defects in the DDR pathway leading to genomic instability contribute to cancer development.
On the other hand, these defects can also present a therapeutic opportunity. A growing number of DDR-targeted drugs have rapidly expanded to include inhibitors of several members involved in the DDR pathway, including PARP, ATM, ATR, CHK1, WEE1, and DNA-PK.
Currently, these inhibitors of DDR components, some of which are in clinical trials, are showing promise. Additionally, new evidence suggests the sensitivity of DDR inhibitors to traditional cancer treatments, as well as the correlation between DDR pathways and immune checkpoint inhibitor (ICI) responses, is driving combination therapies based on DDR inhibitors. Drugs targeting the DNA repair pathway are increasingly showing their importance in the field of cancer treatment.
DNA Damage and DNA Damage Response
To maintain genome integrity, a complex DNA repair system is employed to combat various forms of DNA damage, a process known as the DNA damage response (DDR). The DDR pathway is divided into three functionally interwoven parts: sensors that detect DNA damage, signal transducers that trigger signal cascades, and effectors that impede DNA repair. These pathways are not mutually exclusive processes but rather coordinate to form a precise regulatory network for DNA repair.
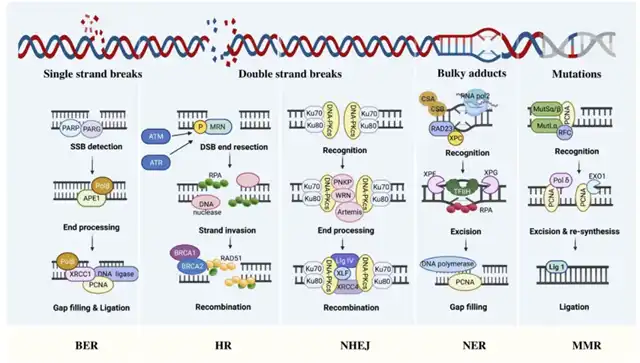
Base Excision Repair (BER) and Nucleotide Excision Repair (NER)
The genome of all organisms undergoes subtle changes due to various endogenously generated genotoxins, such as reactive oxygen species (ROS), ionizing radiation, and alkylating agents. Most minor changes in DNA, such as single-strand breaks (SSBs), are repaired by the BER pathway.
BER is initiated by damaged bases, which are then excised and replaced with newly synthesized DNA. Subsequently, the apurinic/apyrimidinic endonuclease (APE) cleaves the AP site, creating a 3′OH end at the damage site. Finally, DNA polymerase and DNA ligase are recruited to the nucleotide gap created by the removal of the damaged base, thus sealing the gap. BER is responsible for repairing minor damage, while larger double-strand breaks (DSBs) that distort the DNA helical structure require the NER pathway for repair. The NER mechanism involves a key protein, excision repair cross-complementing protein 1 (ERCC1), which clears the DNA near the break and then replaces it with normal DNA replication.
Homologous Recombination (HR) and Non-Homologous End Joining (NHEJ) In mammalian cells, HR and NHEJ are the two main pathways for repairing DSBs. Because sister chromatids are required as templates for new DNA synthesis, the HR pathway can repair DSBs in the S/G2 cell cycle phases, while NHEJ is active in all cell cycle phases except M phase.
HR analyzes homologous sequences from other parts of the genome, gathering information lost at the break site. HR first trims the broken ends, then forms Rad51 nucleoprotein filaments with Brca2, initiating the search for homologous sequences and facilitating the formation of connection molecules between the broken DNA and the homologous template, completing the repair.
NHEJ is simpler than HR, directly rejoining the broken ends. The basic factors required for NHEJ are a heterodimer composed of Ku70/Ku80 and the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs), which recognize DSBs and promote downstream signaling factors for NHEJ, such as XRCC4, XLF, and DNA ligase IV. While the NHEJ mechanism is simpler, it can sometimes lead to rearrangements, whereas HR is thought not to introduce errors.
In addition to HR and NHEJ, there is a DSB repair pathway that shares similar mechanisms with these two main DSB repair pathways but is genetically distinct, known as the alternative end joining (a-EJ) pathway. The a-EJ pathway can share a similar initial process with HR or form a DNA end-joining factor without a homologous template like NHEJ. Increasingly, research is focusing on the a-EJ pathway as a potential therapeutic target for NHEJ or HR-defective cancer cells.
Mismatch Repair (MMR)
In addition to damage from exposure to genotoxins, DNA damage can also result from aberrant DNA processing. The pathway that repairs replication-related errors in DNA is called MMR. During DNA synthesis, MMR corrects nucleotide errors integrated, preventing permanent DNA changes in dividing cells. Therefore, MMR defects caused by gene mutations or epigenetic silencing may increase the spontaneous mutation rate, often associated with hereditary and sporadic cancers.
Translesion Synthesis and Template Switching
DNA damage tolerance (DDT) serves as a fundamental bypass mechanism for repairing replication-stalling DNA damage, allowing DNA replication to proceed past obstructive elements. Translesion synthesis (TLS) is one of two distinct DDT modes, depending on the function of a specialized TLS polymerase rather than the replicative DNA polymerase, which can directly replicate across the damaged site. Due to the limited proofreading activity of TLS polymerases, the TLS mechanism is error-prone, increasing the risk of mutations. Not surprisingly, TLS is a major source of cellular mutations.
In contrast, another mode of DDT, termed template switching (TS), involves recombination onto a homologous DNA template on the sister chromatid, similar to the HR process, and is considered more accurate than TLS. The repair activities of TLS and TS begin after the replication fork, indicating that they may occur during or after DNA replication, with TS starting in early S phase and TLS starting in late S phase.
Fanconi Anemia (FA) Pathway
Fanconi anemia is a rare genetic disease caused by biallelic mutations in Fanconi genes, and affected patients have a reduced response to DNA damage. Fanconi anemia has been identified as a DNA repair pathway that can clear barriers to DNA replication and transcription, namely DNA interstrand crosslinks (ICLs).
ICLs can be formed by aldehydes during various metabolic reactions (such as lipid peroxidation and ethanol metabolism) and chemotherapy (such as platinum). While intrastrand crosslinks are repaired through the NER pathway, ICLs are mainly repaired through the FA pathway. After the detection of ICLs by the UHRF1 protein and the FANCM-MHF1-MHF2 complex, the FA core complex is recruited to the chromatin and monoubiquitinates the substrate FANCI and FANCD2. Ubiquitinated FANCD2-I recruits various DNA endonucleases to cleave at the crosslink site, allowing for the recognition of the crosslinked DNA and the removal of nucleotides, thereby generating DNA substrates suitable for recombinational repair.
O6-Methylguanine-DNA Methyltransferase Pathway
It is well known that DNA methylating agents can inhibit DNA methylation and produce a wide range of DNA adducts, such as O6-methylguanine (O6MeG) and O4-methylthymine, which can lead to base mismatches and subsequent point mutations. Among these, O6MeG is considered a major premutagenic lesion and is repaired by O6-methylguanine-DNA methyltransferase (MGMT).
MGMT removes the methyl group from O6MeG, irreversibly transferring it to a cysteine residue in the active site of MGMT and restoring the guanine base. Since MGMT repairs O6MeG, it is a key factor in preventing G:C to A:T transition mutations. However, in the process, MGMT is inactivated by losing its active-site cysteine residue, which is replaced by a methyl group, rendering MGMT unable to bind to the DNA for further repair. Due to this inactivation mechanism, MGMT has only a limited capacity to repair O6MeG lesions.
DNA Repair Pathway Targeted Therapies in Cancer
The importance of DNA repair pathways in cancer treatment is increasingly evident, with DNA repair-defective cancers being particularly susceptible to DNA-damaging agents. Targeted therapies that exploit DNA repair deficiencies are showing promise in clinical trials and are becoming a focus of research in cancer treatment.
Poly (ADP-Ribose) Polymerase (PARP) Inhibitors PARP inhibitors have emerged as a promising class of targeted cancer therapies, particularly in tumors with BRCA1/2 mutations. PARP enzymes are involved in the repair of SSBs, and PARP inhibitors block the repair of these SSBs, leading to the accumulation of DSBs and ultimately cell death. While initially effective in BRCA-mutant tumors, PARP inhibitors are now being investigated in various cancers, including prostate, pancreatic, and ovarian cancers, either alone or in combination with other agents.
ATR Inhibitors
ATR (ataxia-telangiectasia and Rad3-related protein) is a key regulator of the DDR pathway, particularly in response to replication stress. ATR inhibitors have shown promise in preclinical studies, particularly in combination with other DNA-damaging agents or checkpoint inhibitors. Clinical trials are ongoing to evaluate the efficacy of ATR inhibitors in various cancers.
CHK1 Inhibitors CHK1 is another key regulator of the DDR pathway, particularly in response to replication stress and DNA damage. CHK1 inhibitors have shown promise in preclinical studies and are being evaluated in clinical trials, either alone or in combination with other agents, in various cancers.
WEE1 Inhibitors
WEE1 is a key regulator of the G2 checkpoint in the cell cycle, allowing cells to repair DNA damage before entering mitosis. WEE1 inhibitors have shown promise in preclinical studies, particularly in combination with DNA-damaging agents or checkpoint inhibitors. Clinical trials are ongoing to evaluate the efficacy of WEE1 inhibitors in various cancers.
DNA-PK Inhibitors
DNA-dependent protein kinase (DNA-PK) is a key regulator of the NHEJ pathway, particularly in response to DSBs. DNA-PK inhibitors have shown promise in preclinical studies and are being evaluated in clinical trials, either alone or in combination with other agents, in various cancers.
Combination Therapies
Given the complexity of the DDR pathway and the crosstalk between different repair pathways, combination therapies targeting multiple components of the DDR pathway are being investigated. These combination therapies aim to exploit synthetic lethality in DDR-defective cancers and overcome resistance to single-agent therapies.
Conclusion
The DDR pathway plays a crucial role in maintaining genome integrity and preventing cancer development. Defects in the DDR pathway can lead to genomic instability and cancer development.
Targeted therapies that exploit DNA repair deficiencies are showing promise in cancer treatment, particularly in DDR-defective cancers.
Future research is needed to further elucidate the role of the DDR pathway in cancer development and identify novel targeted therapies.
Targeted DNA Repair Pathway in Cancer Treatment
Reference:
1. Targeting DNA repair pathway in cancer:Mechanisms and clinical application. MedComm (2020). 2021 Dec; 2(4):654–691.
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.



