Infections in adult and pediatric patients receiving CAR-T cell therapy
- Aspirin: Study Finds Greater Benefits for These Colorectal Cancer Patients
- Cancer Can Occur Without Genetic Mutations?
- Statins Lower Blood Lipids: How Long is a Course?
- Warning: Smartwatch Blood Sugar Measurement Deemed Dangerous
- Mifepristone: A Safe and Effective Abortion Option Amidst Controversy
- Asbestos Detected in Buildings Damaged in Ukraine: Analyzed by Japanese Company
Infections in adult and pediatric patients receiving CAR-T cell therapy
- Red Yeast Rice Scare Grips Japan: Over 114 Hospitalized and 5 Deaths
- Long COVID Brain Fog: Blood-Brain Barrier Damage and Persistent Inflammation
- FDA has mandated a top-level black box warning for all marketed CAR-T therapies
- Can people with high blood pressure eat peanuts?
- What is the difference between dopamine and dobutamine?
- How long can the patient live after heart stent surgery?
Infections in adult and pediatric patients receiving CAR-T cell therapy
Introduction
Chimeric antigen receptor T cell therapy (CAR-T cell therapy) is a new type of adoptive immunotherapy, which represents a huge advancement in cancer therapy.
T lymphocytes are genetically engineered using viral vectors carrying chimeric antigen receptors (CAR).
CAR encodes an extracellular domain for recognizing tumor antigens (for example, recognizing B cell CD19), which is connected to an intracellular signal domain that mediates T cell activation.
These modified T lymphocytes are then injected into the patient’s body to use the patient’s immune system to target and eliminate cancer cells.
Early clinical trials have shown a high response rate in relapsed and/or refractory (R/R) B cell malignancies. It is worth noting that some of the reactions are long-lasting, indicating the potential for healing.
These results prompted the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) to accelerate the approval of two CAR-T products (tisagenlecleucel and axiabtagene ciloleucel) for the treatment of children and children’s R/RB cell hematological malignancies. Adult patients: acute lymphoblastic leukemia (ALL) and diffuse large B-cell lymphoma (DLBCL).
The early prospects of these therapies have led to a surge in clinical trials of CAR-T therapies, with more than 1,000 internationally registered trials involving a wide range of different manufacturing technologies, antigen targets, and cancer types.
With the rapid spread of research and the accelerated development of CAR-T cell therapy, people still lack the understanding of infection-specific toxicity.
The recommendations are mainly driven by knowledge inferred from other hematology treatment settings, such as autologous and allogeneic hematopoietic cell transplantation (HCT).
The purpose of this review is to summarize the current literature on infections in adult and pediatric patients receiving CAR-T cell therapy, and to make recommendations to prevent infections.
Mechanisms to reduce immunity
The increased risk of infection in patients receiving CAR-T cell therapy is multifactorial. Hematological malignancies inherently cause immune dysfunction.
At present, CAR-T cell therapy is only approved for patients with R/R diseases. Therefore, the patient has received multiple treatments before, leading to cumulative immune dysfunction.
In a study of adult patients with R/R lymphoma, 52% had previously received more than three treatments, and 49% had previously received autologous HCT.
Among ALL patients, the previous rate of allogeneic HCT was very high, with the Seattle cohort reporting rate of 55%. The retrospective cohort has determined that the median is 3-4 previous treatments, 36-38% of previous SCTs.
In other malignancies, the previous treatment line is a known risk factor for infection, especially invasive fungal infections.
Another factor leading to the risk of infection is the use of lymphocyte depletion chemotherapy before CAR-T infusion to deplete T regulatory cells and other immune cells, thereby enhancing T cell function and improving the anti-tumor efficacy of the transferred CAR-T. To date, most trials have used a combination of fludarabine and cyclophosphamide.
The resulting loss of mucosal integrity and decreased blood cells increases the risk of early infection, as does the longer-term lymphotoxic effects of purine analogs (such as fludarabine).
The use of immunosuppressive therapy to treat the complications of CAR-T cell therapy may further increase the risk of infection.
The risk of the unique toxicity of cytokine release syndrome (CRS) or immune effector cell-related nervous system syndrome (ICANS) varies depending on the CAR-T cell product used, disease, and patient-related characteristics.
The European Medicines Agency’s review of tisagenlecleucel found that the incidence of CRS was 58-80%, while the incidence of ICANS was 21-38%.
Depending on the severity of these complications, it is necessary to use tocilizumab (IL-6 receptor monoclonal antibody) and/or high-dose corticosteroids to treat such complications.
Commercial CAR-T products currently target B cell markers, such as CD19, so as to have effects on malignant B cells and normal circulating B cells.
The long-lasting response of this “living drug” may mean that B-cell aplasia lasts for months to years.
B-cell dysplasia and subsequent hypogammaglobulinemia will bring additional risks of infection because of reduced humoral immunity, especially against invasive bacterial infections and viral infections.
The last factor that is gaining more and more attention is long-term cytopenia. Due to the bone marrow toxicity of chemotherapy, early neutropenia is both common and predictable.
Several studies following the initial trial have determined that a large proportion of patients have prolonged advanced cytopenia.
A phase 1b/2 trial conducted in Israel found that 76% of patients who responded to CAR-T cell therapy developed neutropenia after the 21st day.
Long-term follow-up of the ZUMA-1 trial cohort found that 11% of patients had prolonged neutropenia for more than 3 months, and a similar late-effect cohort that was tested in Seattle found that 16% of patients had cytopenias 21 months after the infusion Symptoms persist.
The mechanism of these cytopenias is not yet clear, but it exceeded the expectations of chemotherapy and continued well after CRS subsided.
Infection rate after CAR-T cell therapy
So far, five studies (four for adults and one for children) have specifically investigated the infection rate of patients receiving CAR-T treatment.
Four studies are retrospective cohort studies of patients participating in CAR-T clinical trials at a single institution, and one study is a study of patients receiving commercial FDA-approved CAR-T cell therapy at a single institution.
Overall, the infection rate after CAR-T cell treatment seems to be comparable to the infection rate of similarly large numbers of pretreated patients.
In these 5 cohorts, the incidence of early infection (<30 days) was 17% to 42%, and the incidence of late infection (30-120 days) was 14% to 31%.
Most reported infections are mild to moderate in severity, with early bacterial infections being more common, and viral infections more common after 30 days.
In general, there are few reports of invasive fungal infections, with the early and late incidence rates as high as 8% and 3%, respectively.
The largest study conducted by Hill et al. studied 133 adult patients who were treated in a mixed population of NHL, CLL, and ALL in the Phase 1/2 open-label trial of CD19 CAR-T cell therapy.
In the first 28 days after CAR-T cell therapy, 30 (23%) patients had 43 infections, and the infection density was 1.19 infections per 100 days (calculated based on the number of infections per patient per 100 days).
On days 29-90 after infusion, the infection density is low, with a risk of 0.67 per 100 days. In the first 28 days, bacterial infections were the most common, occurring in 16.5% of patients, and about half of them were due to bacteremia. The second is viral infection, which occurs in 8% of patients, and the second is fungal infection, which occurs in 3% of patients.
After 28 days, viral infections were the most common, followed by bacterial infections. Most infections (50%) were of mild to moderate severity, and there were 2 deaths related to infection; one was delayed infection (90 days after infusion), and tracheobronchitis caused by Aspergillus flavus was the main cause of death One is that severe Clostridium difficile is a secondary cause of death in the case of fatal CRS.
After 90 days of infusion, the infection rate remained stable, with a density of 0.55 per 100 days.
The same cohort was followed up for long-term. Most infections are still mild and involve the upper respiratory tract, 80% of which are treated in an outpatient setting.
At the Memorial Sloan Kettering Cancer Center (MSKCC), a study of 53 adult patients reported a similar pattern, who participated in a phase 1 trial of CD19 CAR T-cell therapy for R/R ALL.
Bacterial infections are most common in the early stages (30% of patients), while viruses dominate in late infections (day 31 to day 180).
The overall fungal infection occurred in 8% of patients in the early stage and 3% in the late stage. There are no reported data on the severity of the infection; however, 3 infection-related deaths occurred after the first 30 days (1 due to multi-drug resistant bacteria and 2 due to multiple microbial sepsis).
A different study from the same center that evaluated patients with R/R DLBCL also noted that bacteria are predominant in early (first 30 days) and late infections (30 to 1 day after infusion).
Urinary tract infection is the most common site of late infection. In this study, the overall infections were mild to moderate (71%), of which 23% were reported as severe infections, 1% were life-threatening, and 1% were fatal.
In a heterogeneous group of 109 patients, participated in three clinical trials of CAR-T therapy (CD19/22 for R/RB cell malignancies; CAR-BCMA for multiple myeloma (B Cell maturation antigen) therapy; and CD19/22 CAR-T) R/RB cell malignant tumors after autologous SCT), 17% of patients experienced early infections, mainly bacterial pathogens, within the first 30 days of infusion.
More than half of the infections identified in this study were grade 4-5 (n=11), and 5 patients died of the infection.
All grades 4-5 infections are caused by bacterial pathogens, and most of them occur within 2 weeks of CAR-T cell therapy.
In this cohort, the rate of severe/life-threatening infections was the highest, which may reflect the local epidemiology, and many multi-drug resistant bacteria were found.
The infection rate in pediatric patients seems to be similar to that in adults.
Vora et al. described the complications of infection in 83 children who received CAR-T cell therapy within three time periods; 90 days before CAR-T, 0-28 days after CAR-T, and 29-90 days after CAR-T .
Before CAR-T cell therapy, 54% of patients developed infections at a risk of 1.23 every 100 days. A high infection rate was observed early after the infusion (days 0-28); 40% of infections occurred (risk density of 2.89 per 100 days), of which bacterial infections were the main one (54%), followed by viral infections (38%) .
Within 29-90 days after infusion, which is dominated by viral infections, the infection density decreases to 0.55 risk per 100 days. In terms of severity, 43% were mild to moderate, 45% were severe, and 13% were life-threatening.
Most serious/life-threatening infections are caused by bacterial pathogens. The only fatal infection occurred on the 51st day after CAR-T cell treatment, which was due to septic shock caused by Aeromonas hydrophila infection.
Other sources of infection-related data come from key trials and drug review evaluations.
Three key trials of CAR-T cell therapy reported adverse events, but detailed information about the infection was often lacking.
The JULIET trial is a phase II international trial of tisagenlecleucel in the treatment of adult relapsed or refractory DLBCL.
In the first 8 weeks after CAR-T cell infusion, 34% of patients developed infections, most of which were grade 3 infections.
The late infection rate is similar to the early infection rate. The ZUMA-1 trial using axicabtagene ciloleucel in refractory DLBCL did not specifically report infection rates, but 85% of patients developed fever, of which 35% was defined as febrile neutropenia.
However, a long-term safety analysis of the ZUMA-1 cohort found that 28% of patients had grade 3 or higher infections.
The Tisagenlecleucel ELIANA trial in children and young adults with B-cell lymphocytic leukemia found that 43% of patients developed infections within the first 8 weeks (mostly grade 3).
Severe infections have been recorded as human herpes virus 6 (HHV-6) encephalitis and systemic mycosis, both of which are associated with long-term neutropenia.
The EMA’s review report on tisagenlecleucel stated that 54% of adults with DLBCL were infected.
Due to the heterogeneity of the trial population, it is difficult to compare the infection rates of different treatment modalities.
A single-arm controlled trial of patients with relapsed/refractory NHL who received CAR-T cell therapy found that the incidence of infection was 13%, while the incidence of infection in patients who received autologous HCT during the same period was 40%.
The infection rate is also better than that of patients receiving allogeneic HCT treatment.
A large registration study of infection complications after allogeneic HCT found that the density of infections that occurred during the first 100 days was 1.72 to 2.06 infections per 100 days, depending on the source of the cells.
Overall, the infection rate after CAR-T cell therapy seems to be comparable to similar therapy for patients who have undergone extensive pretreatment.
Risk factors associated with infection
The four previously mentioned studies investigated factors related to the increased risk of infection in their cohort.
Hill et al. adjusted the multivariate model of baseline characteristics and found that the diagnosis of ALL, receiving more than 4 previous treatments, and receiving high-dose CAR-T cells (2x 10^ 7 cells per kilogram ) were associated with higher densities Infection rate in the first 28 days.
In a multivariate analysis, the severity of cytokine release syndrome (CRS) is the only treatment-related factor that is associated with an increased risk of infection.
Each increase in the CRS level increases the risk of infection by 3.4 (0 vs. 1-3 vs. 4- 5).
Similarly, Park et al. found that CRS (≥ grade 3) was independently associated with an increased risk of infection (adjusted hazard ratio 2.67; p=0.05), especially bloodstream infection (adjusted HR 19.97; p<0.001). Vora et al. performed a Cox proportional hazard model on variables that affect the risk of infection, and found that prior SCT (HR 2.15; 95% CI 0.98–4.75) and hypogammaglobulinemia after treatment (HR 2.41; 95% CI 1.02) –5.69) The risk of infection in the first 28 days after CAR-T cell therapy is related to the increased risk of infection.
In a univariate analysis, Wudhikarn et al. found that impaired baseline physical status, occurrence of ICANS ≥ 2 and systemic corticosteroid use were associated with an increased incidence of infection.
However, after multivariate analysis, only systemic corticosteroid use remained a risk factor (HR 2.22; 95% CI 1.05–4.67).
Septicemia
One of the major challenges in the early stage after CAR-T cell therapy is to distinguish CRS from sepsis.
The main characteristics of CRS are fever and hemodynamic instability. CRS usually occurs in the first week after CAR-T cell infusion, which coincides with the risk of neutropenia and bacterial sepsis.
In addition, an increase in the severity of CRS is related to an increase in the risk of infection.
Although the mechanism of this association is unclear. Severe CRS is characterized by endothelial activation and increased blood angiogenin 2 and von Willebrand factor.
It is reasonable that CRS-induced endothelial injury may initiate or promote the infection process.
To help distinguish between sepsis and CRS, researchers are trying to determine which biomarkers can be used as predictive tools.
Park et al. identified cytokines related to CRS (interferon gamma (IFN- gamma), tumor necrosis factor alpha (TNF-alpha), interleukin (IL)-6, IL-10 and IL-15), but No differences in cytokine levels were found between CRS co-infected patients and CRS patients alone.
Luo et al. found that there are differences in the inflammatory characteristics between CRS (elevated ferritin and IL-6) and infection (elevated IL-6, ferritin levels within the normal range). They also found that in a set of cytokine studies, IL-8, IL-1β, and IFN-γ have good sensitivity and specificity for identifying grade 4-5 infections.
Blood-borne virus
Due to concerns about viral reactivation and fulminant hepatitis, patients with human immunodeficiency virus (HIV), hepatitis B (HBV) or hepatitis C (HCV) are usually excluded from CAR-T cell therapy trials.
Due to the advancement in the care of patients with blood-borne viruses, it is more and more common for patients with chronic viral infections to have hematological malignancies.
The safety of CAR-T cell therapy in these patients is mainly limited to case reports.
(1) Hepatitis B (HBV)
Existing data show that CAR-T therapy is safe for HBV patients, provided that they have received entecavir and other targeted antiviral therapy.
Although HBV reactivation, including fulminant hepatitis, and deaths are rarely reported, all occurred in patients who discontinued entecavir.
The largest data set of patients with combined HBV and B-cell carcinoma receiving CAR-T cell therapy comes from a cohort study in China.
They identified 12 patients with chronic HBV (surface antigen positive) who were receiving antiviral therapy, and 29 patients who had cured HBV (surface antigen negative, core antibody positive) and received regular HBV DNA monitoring.
The safety and effectiveness of CAR-T cell therapy were compared with 29 patients who were not infected with HBV.
Three patients experienced HBV reactivation after receiving CAR-T cell therapy, two had chronic HBV, and one had cured HBV (subsequent to entecavir treatment). None of the patients experienced HBV-related hepatitis episodes, and one patient’s alanine aminotransferase (ALT) was elevated by grade 3.
There were no significant differences in toxicity or response related to CAR-T cell therapy between the HBV and non-HBV cohorts.
A much smaller retrospective cohort study also conducted in China identified three patients with both B-ALL and HBV.
All patients have chronic HBV and are receiving antiviral therapy. During the follow-up period, no patients experienced HBV reactivation.
They further explored the effect of HBV positive on the anti-tumor efficacy of manufactured CAR-T cells through in vitro and in vitro models, and found the same effective tumor eradication efficacy.
(2) Human immunodeficiency virus (HIV)
Two patients with acquired immunodeficiency syndrome (AIDS)-related B-cell lymphoma received CAR-T cell therapy at Massachusetts General Hospital.
The first patient had previously refused antiretroviral therapy (ART). When evaluating CAR-T cell therapy, the CD4 count was 52 cells/mm3 and the viral load was 643,000 copies/ml.
He was evaluated by a multidisciplinary team and agreed to adhere to antiretroviral therapy. He successfully received CAR-T cell therapy.
At the last follow-up, he still insisted on receiving ART, his viral load was undetectable, and his lymphoma was in complete remission.
In the second case, HIV was controlled by ART during the diagnosis of lymphoma, and he successfully received CAR-T cell therapy without significant toxicity.
Interestingly, the production of CAR-T cells uses retroviruses (including lentivirus) for gene transfer, causing the components of retroviruses to become a permanent part of the injected CAR-T cell products.
Therefore, there are reports that patients who have received such products have false positive HIV nucleic acid tests.
Retroviruses cannot replicate and therefore cannot produce new retroviruses, but the impact of false positive tests may cause delays in cancer treatment and family pressure.
For blood-borne virus patients, these case reports indicate that despite active viral infections, CAR-T products can still be produced, they can be safely used with antiviral therapy, and they can induce lasting remission without significant Increase toxicity.
The involvement of infectious disease experts, ensuring appropriate treatment and monitoring, and the inclusion of future trials are essential to improve results and understand the complexity of patients with chronic viral infections.
Reactivation of latent DNA viruses
Overall, there is little evidence of clinically significant reactivation of latent DNA viruses after CAR-T cell therapy.
There are few data on the rate of viral reactivation in CAR-T cell therapy.
Given the high pretreatment rate of CAR-T cell recipients (including approximately one-third of receiving previous allogeneic HCT), it can be expected that the cytomegalovirus (CMV) reactivation rate will be significant. In the above-mentioned cohort study, there was no routine screening for CMV and EBV, and the detection was clinically driven.
Park et al. reported no CMV or EBV, Hill et al. reported 4 cases of viremia and only 1 case of end organ disease; Luo et al. reported 3 cases of DNA/RNA high-throughput detection of CMV virus without end organs -Generation sequencing of disease records;
Wudhikarn et al. found 3 cases of CMV viremia without end-organ disease; and Vora et al. reported no CMV or EBV in the pediatric population. There is a case report of progressive multifocal encephalopathy after CAR-T cell therapy.
Invasive fungal infection IFI
Due to differences in baseline epidemiology and antifungal preventive measures, comparing the incidence of invasive fungal infection (IFI) between studies is challenging.
The early landmark trial of tisagenlecleucel in refractory ALL reported an IFI rate of 13%, while in refractory NHL patients treated with axicabtagene ciloleucel, the reported incidence was 5%.
The Hill cohort reported that among people receiving fluconazole prophylaxis, the IFI breakthrough rate was 2%.
Park et al. reported a 7% IFI breakthrough rate. All affected patients received micafungin prophylaxis and were diagnosed with neutropenia at the time of diagnosis.
The Wudhikarn cohort was from the same center as the Park cohort (MSKCC), and it was noted that the cumulative incidence of IFI in 1 year was 4%. In general, in the published IFI report after CAR-T cell therapy, infection occurs in the early and late stages after treatment, and most patients suffer from CRS and neutropenia.
However, using this information to guide prevention strategies is challenging because most CAR-T treated patients will develop neutropenia and CRS, but only a few will develop IFI.
The current IDSA guidelines for the active prevention of mold in hematological malignancies recommend active prevention of mold when the incidence of IFI is >6%.
Some argue that this recommendation should be extrapolated to CAR-T cell patients whose baseline IFI incidence is known.
If the baseline incidence is <6%, then risk stratification based on traditional risk factors (neutropenia and corticosteroid use) is recommended.
This is a similar approach recommended in a recent review. It is recommended to identify high-risk patients and consider active prevention of mold in these patients (usually those receiving tocilizumab, high-dose steroids, second-line immunosuppressive agents for CRS and/or ICANS Patients, and severe neutropenia).
Others suggest an individualized approach, using severe CRS as a risk factor that triggers the use of active mold prevention.
Precautions to prevent infection
(1) Antibiotic prevention
Antimicrobial prevention is the main method to prevent infections in immunocompromised hosts, but according to epidemiology, institutional guidelines and individual clinician practices, the methods used are different worldwide.
Therefore, the schemes used in CAR-T cell therapy are also very different.
In the Park et al. cohort, 87% received PJP prophylaxis (60% trimethoprim-sulfamethoxazole (TMP SMX), 17% pentamidine), and 98% received antiviral prophylaxis (72% acyclovir , 21% valacyclovir), 79% received antifungal prophylaxis (60% micafungin, 11% voriconazole) and no patients received antibacterial prophylaxis.
In contrast, the Seattle cohort used a method that mimics the autologous HCT antibacterial prevention program.
Patients received levofloxacin and fluconazole until ANC>500 cells/mm3, valacyclovir or acyclovir and TMP-SMX until >3 months after CAR-T cell therapy.
As an example of differences in local epidemiology and guidelines, the Luo cohort in Wuhan received imipenem-cilastatin, teicoplanin, linezolid, and voriconazole during neutropenia.
The Wudhikarn cohort comes from a non-clinical trial setting.
For most studies, there are no formal institutional guidelines for antibiotic prevention. Three months before the end of the cohort study, MSKCC implemented standardized guidelines, including acyclovir for the prevention of herpes simplex virus (HSV), fluconazole for antifungal prevention, and TMP-SMX for PJP prevention .
A survey of professional pharmacists participating in CAR-T cell therapy found that respondents reported widespread use of antiviral and pneumocystis prevention, as well as a high use rate (~90%) for antifungal and bacterial prevention.

(2) Immunoglobulin replacement
Infection is a sign of primary immunodeficiency, especially invasive bacterial infections and bacteremia, as well as viral respiratory infections.
As mentioned above, the persistent B cell hypoplasia associated with CAR-T cell therapy and the subsequent hypogammaglobulinemia can impair humoral immunity and may bring the same risk of infection as primary immunodeficiency.
In view of the lack of clinical trial data, the use of immunoglobulin substitutes to change this risk is controversial. Current management practices vary widely and are well described in recent reviews.
Practical recommendations from the practice of SCT and other immunodeficiency states suggest that if severe hypogammaglobulinemia (IgG<400mg/dL) or moderate hypogammaglobulinemia (400-600mg/dL), in CAR-T cells In the first three months after treatment, consider replacing IgG for repeated and severe infections.
(3) Growth factor support
The role of granulocyte macrophage colony stimulating factor (GM-CSF) and granulocyte colony stimulating factor (G-CSF) in CAR-T cell therapy is unclear.
The manufacturer’s product information does not recommend the use of growth factor support within the first three weeks after CAR-T cell infusion or before CRS is resolved.
It has been found that the levels of GM-CSF and G-CSF in patients with severe neurotoxicity are elevated.
In vivo studies have found that GM-CSF plays a key role in promoting CRS, and the artificial inactivation of GM-CSF reduces the biomarkers of macrophages.
However, long-term cytopenias after CAR-T cell therapy are both common and severe. In this case, the use of growth factor support has been widely practiced.
A single-center study compared patients who did not receive G-CSF therapy with patients who received preventive G-CSF therapy from the 5th day after CAR T cell therapy, and found that the risk of severe CRS or ICANS did not increase.
Further research is needed to understand the optimal timing and duration of growth factor support.
(4) Vaccination
The long-term effects of CAR-T cell therapy on humoral immunity are unclear. Terminally differentiated B cells (such as plasma cells) have low CD19 expression and may not be affected by CAR-T cell therapy, thus keeping the established humoral immunity intact.
A small study of 16 patients who participated in the CD19 CAR-T cell clinical trial showed that despite the hypoplasia of B cells, plasma cells in the bone marrow were retained [47].
Another study conducted in a clinical trial on 39 patients receiving CAR-T cell therapy found that the average serum total IgG concentration decreased slightly (about 100mg/dL) after CAR-T cell therapy, but the average serum measles specificity Similar IgG levels before and after treatment indicate that pathogen-specific immunity may be preserved [48].
CD19 B cell counts, serum IgG, IgM, and IgA levels, pathogen-specific IgG levels, and vaccine response may help guide decisions about humoral immunity.
There is a lack of data on vaccination strategies in CAR-T cell therapy, and the role of vaccines is unclear.
The patient population is heterogeneous; children may have fewer established plasma cell clones and may not have completed their primary vaccine series, patients who have previously received HCT may be at different stages of re-vaccination, patients who have received regular IgG replacement, and have undergone A large number of pretreated patients have B cell hypoplasia.
The immunogenicity and safety of vaccination in this diverse population are still unclear, and further research is needed to guide recommendations.
Suggestion
As the commercial availability of CAR-T cell products continues to increase, a number of guidelines and documents have been issued that contain recommendations on selection, delivery of CAR-T cell therapy, and toxicity management.
Most infection-related recommendations are inferred from allogeneic and autologous SCT data.
The three large-scale social recognition documents published in 2019 differ slightly in antimicrobial prevention recommendations, especially in terms of antibacterial and antifungal prevention.
The US guidelines advocate the use of fluoroquinolones for prevention during neutropenia.
These differences reflect the lack of data on the effectiveness and safety of fluoroquinolones for prevention in many immunocompromised host populations, as well as differences in local morbidity, resistance patterns and epidemiology.
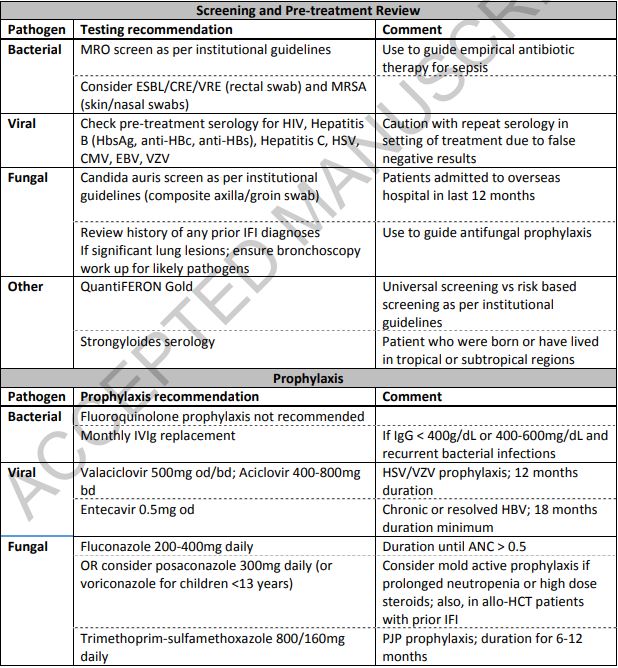
Table 2 represents our recommended antimicrobial prevention and treatment methods for pediatric and adult patients based on this review, focusing on infectious disease consultation, pre-treatment screening, and individualized patient methods.
It is also important to have a deeper understanding of immune reconstitution after CAR-T cell therapy.
Patient and disease characteristics before treatment, coupled with the unpredictable duration of cytopenias, hypogammaglobulinemia, and B cell hypoplasia after CAR-T cell treatment, lead to significant heterogeneity in the overall state of immune damage.
In this case, clinical infection risk assessment has become more and more complex and challenging.
In the short term, clinical studies to determine the role of immunoglobulin replacement, antibiotic prophylaxis, and vaccination strategies will be important tools for this assessment.
In the long run, quantifying immunosuppression through immunoassays to predict risk and personalized infection prevention strategies may be an integral part of effective and efficient supportive care.
Reference:
CAR-T cell therapy and infection: a review
doi:10.1080/14787210.2021.1855143
Infections in adult and pediatric patients receiving CAR-T cell therapy
(source:internet, reference only)
Disclaimer of medicaltrend.org
Important Note: The information provided is for informational purposes only and should not be considered as medical advice.