Gastrointestinal microbes predict peripheral inflammation of tuberculosis
- Normal Liver Cells Found to Promote Cancer Metastasis to the Liver
- Nearly 80% Complete Remission: Breakthrough in ADC Anti-Tumor Treatment
- Vaccination Against Common Diseases May Prevent Dementia!
- New Alzheimer’s Disease (AD) Diagnosis and Staging Criteria
- Breakthrough in Alzheimer’s Disease: New Nasal Spray Halts Cognitive Decline by Targeting Toxic Protein
- Can the Tap Water at the Paris Olympics be Drunk Directly?
Gastrointestinal microbes predict peripheral inflammation of tuberculosis
Gastrointestinal microbes predict peripheral inflammation of tuberculosis. Gastrointestinal microbiota composition predictsperipheral inflammatory state during treatment of human tuberculosis.
Tuberculosis (tuberculosis, TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) infection.
Guide
The composition of gastrointestinal microorganisms can significantly affect the systemic immune response, but its influence on the pathogenesis of infectious diseases and the therapeutic effect of antibiotics is poorly understood. Because it is currently difficult to isolate the immune effects of antibiotic-induced pathogen elimination and related microbial changes, the above-mentioned problems have been seldom studied in the population.
In this study, the researchers separately analyzed two longitudinal study cohorts (35 and 20) of tuberculosis (TB) treatment and a cross-sectional study of 55 healthy controls. In these studies, the researchers collected subjects’ stool samples (used for microbiome analysis), saliva (used to determine the bacterial load of Mycobacterium tuberculosis (Mtb)), and peripheral blood (used for transcriptome analysis). analysis).
In addition, the study separated the microbial effects from pathogen sterilization by comparing standard TB treatments with experimental therapies that did not reduce Mtb load. Random forest regression analysis of the microbiome-transcriptome-saliva sample data from the two longitudinal cohorts showed that the renormalization of TB inflammation and the elimination of Mtb pathogens, the increased abundance of Clusters IV and XIVa Clostridia, and the increase of Bacilli and Proteobacteria Related to reduced abundance.
Peripheral gene expression and microbiome analysis in independent cohorts of healthy individuals showed similar associations. The results of this study indicate that the decrease in pathogen abundance and microbial changes induced by antibiotics are related to the changes in the active TB inflammatory response induced by treatment, and the response to antibiotic treatment may be a combination of pathogen clearance and microbial-driven immune regulation.
Paper ID
- Original name: Gastrointestinal microbiota composition predictsperipheral inflammatory state during treatment of human tuberculosis
- Journal: Nature Communications
- IF: 12.121
- Posting time: 2021.2.18
- Corresponding author: Michael S. Glickman & VanniBucci
- Corresponding Author Unit: Department of Immunology and Pathogenic Microbiology, Cornell Medical College, University of Massachusetts Microbiome Research Center
Experimental design
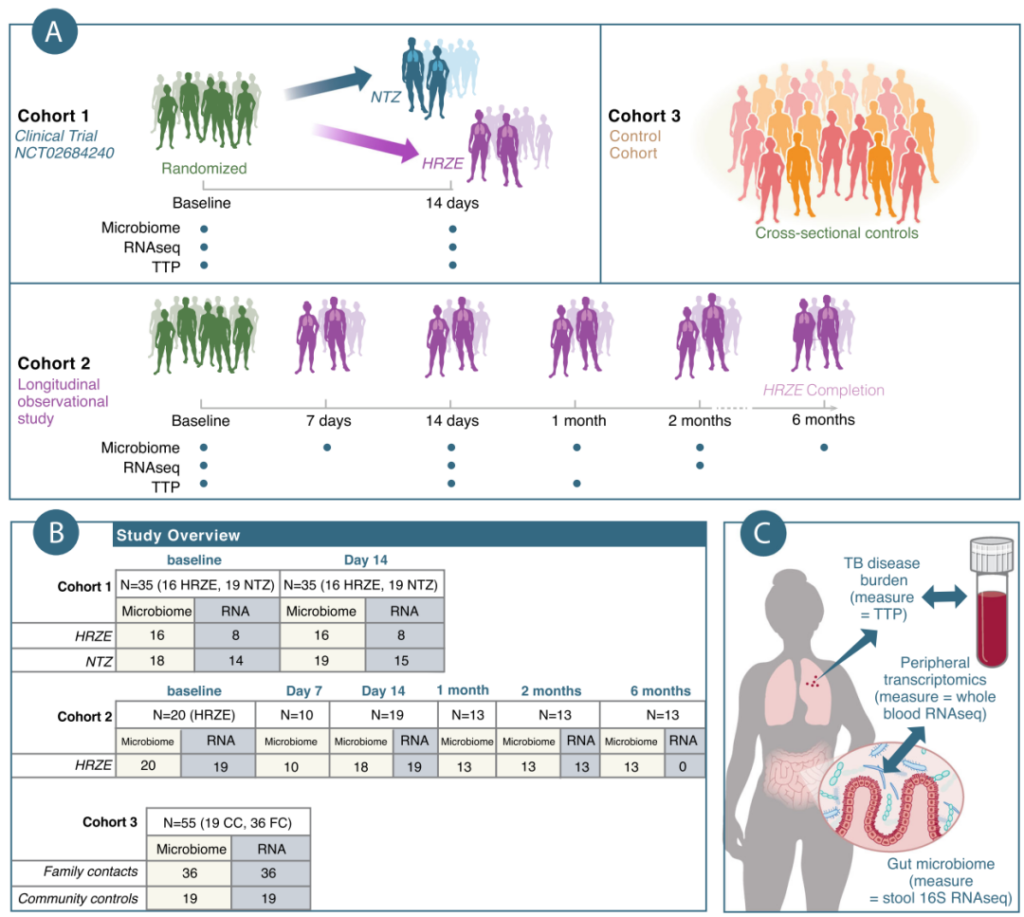
Figure 1 Overview of the experimental design of this study. A. This study investigated the relationship between the microbiome and the transcriptome of three different cohorts from Haiti. Among them, cohort 1: conduct a 2-week longitudinal and interventional clinical trial, including a secondary analysis of randomized clinical trials conducted on research volunteers. On this basis, the researchers collected the bacterial load (TTP) of Mycobacterium tuberculosis in patients with active tuberculosis, fecal microbiome analysis and peripheral transcriptomics analysis, and then randomly divided them into HRZE treatment group (TB standard treatment) or Nitrazoxanide treatment group (Nitazoxanide, NTZ). Cohort 2: Conducted a 6-month longitudinal and observational study. The subjects had been undergoing follow-up studies during the 6-month TB treatment process. At the same time, the subjects’ TTP, fecal microbiome and transcriptome data were collected. Cohort 3: (cross-sectional and observation group) a cross-sectional and observational study composed of individually recruited healthy volunteers, about half of which were family contacts (FC) of healthy and TB-negative active TB patients, The other half are community controls (CC) with no known exposure to tuberculosis. The study also performed fecal microbiome analysis and peripheral transcriptome analysis on the aforementioned individuals. B. The number of individuals included in the different study cohorts of this study. C. Schematic diagram of sample collection and corresponding analysis of subjects included in this research.
Results:
1 After two weeks of HRZE or NTZ treatment, the patient’s gut microbiota diversity was significantly reduced
A prospective, randomized, early bactericidal activity (EBA) study (NCT02684240) was conducted at the GHESKIO Center in Port-au-Prince, Haiti. The object of the study was newly-treated, drug-susceptible and uncomplicated tuberculosis (TB). ) Adult patients. In this study, 30 subjects were randomly divided into two groups. One group was given NTZ treatment, 1000 mg orally twice a day; the other group was given standard oral treatment (HRZE), that is, isoniazid 300 mg per day, Rifal Pyrazinamide was 600 mg per day, pyrazinamide was 25 mg/kg per day, and ethambutol was 15 mg/kg per day for a total of 14 days (Figures 1A and 2A). At the same time, 5 subjects were pre-screened to receive HRZE treatment. The primary endpoint of this test is based on the bacterial load (time to culture positivity, TTP) in the sputum sample in the BACTEC liquid culture system as a microbiological evaluation index. Sputum samples from subjects were collected every other day from 6 pm to 9 am to quantify the activity of Mycobacterium tuberculosis in subjects in each treatment regimen.
Compared with the baseline TTP of the linear mixed-effects model, HRZE resulted in a predictable increase in the subject’s TTP in the first two weeks of treatment (corresponding to a reduced bacterial load), and NTZ was used as the reference level of treatment (Figure 2B). Although NTZ has high activity in vitro, the patient had no significant effect on TTP after 14 days of treatment (p>0.05) (Figure 2B). The reason may be that NTZ fails to penetrate the sputum and thus cannot exert antibacterial effects. Therefore, the relevant subjects were subsequently treated with HRZE standard treatment.
Although the researchers have reported in previous studies that the use of HRZE therapy can eliminate Clostridiales, the cross-sectional design of the relevant studies did not take into account the rapidity of the above-mentioned effects, and most importantly, did not include pretreatment to assess the baseline microbial composition sample. In order to study the changes in the gut microbiome caused by HRZE or NTZ intervention, the study extracted and amplified the DNA of bacteria and archaea in the patient’s gut, and used the V4-V5 region for 16S rDNA sequencing analysis. Stool samples from patients were collected before the start of treatment and on the 14th day of treatment (Figure 1A). Principal coordinate analysis (PCoA) based on the Bray-Curtis distance algorithm showed that the components with the largest changes in the microbiome data qualitatively represent the changes in the intestinal microbiome community structure of patients after two weeks of NTZ treatment (Figure 2C) .
In addition, in order to compare the impact of the two intervention methods on the diversity of the gut microbiome a of patients, the researchers separately counted the Inverse Simpson diversity index of each sample, and used a linear mixed effects model (Diversity~Sex+Age+Batch+ Treatment+Time+Treatment: Time+1| ID) performed a regression analysis on the index. We found no difference in alpha diversity between the two arms at baseline, while both treatments significantly reduced microbial diversity (p<0.01, see Supplementary Data 3), and there was no significant difference between NTZ treatment and HRZE (p >0.05, see Supplementary Data 3) (Figure 1D).
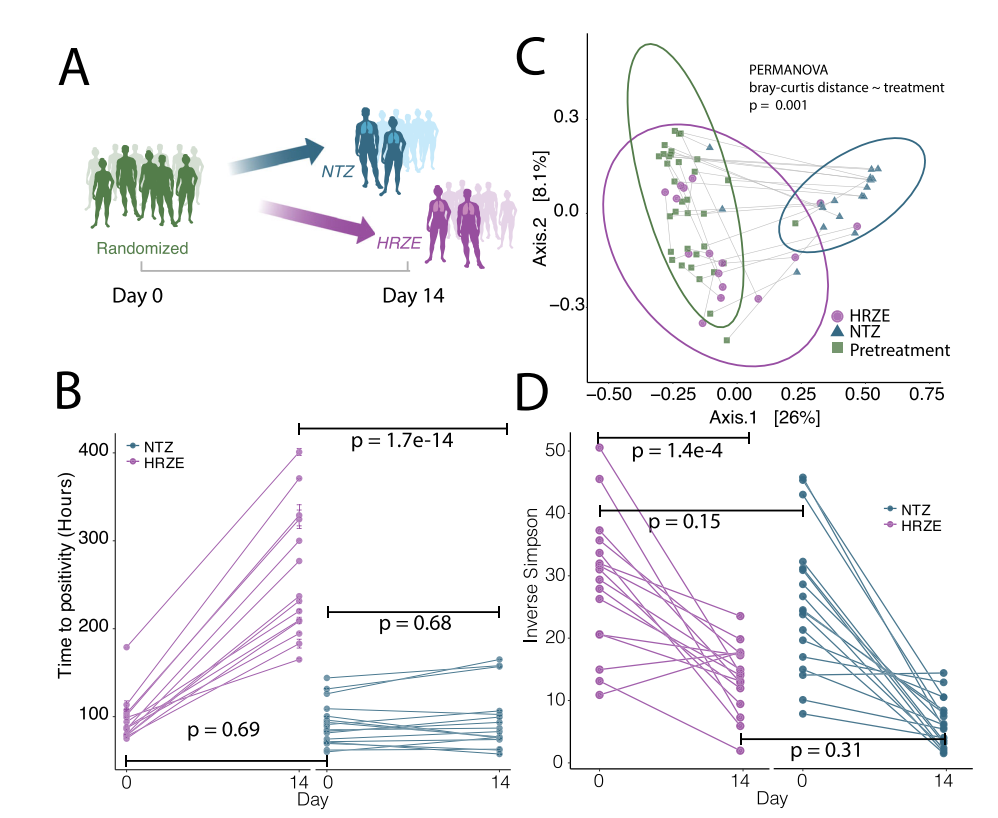
Figure 2 Two weeks after treatment with HRZE or NTZ, the diversity of the gut microbiota was changed. Only HRZE treatment reduced the bacterial load of Mtb. A. Schematic diagram of clinical trials comparing the effect of HRZE and NTZ in removing pathogenic bacteria in vivo. B. For the NTZ treatment group and the HRZE treatment group, the positive reaction time (TTP) of Mycobacterium tuberculosis sputum samples matched on day 0 and day 14 is shown as a range of 2 to 3 technical replicates. Among them, HRZE group: n=16; NTZ group: n=19. A linear mixed-effect model (TTP~1+Sex+Age+ Treatment+Time + Treatment: Time+1|ID) was used to determine the significance of the difference between treatment and pretreatment in each group, where “Treatment” means NTZ treatment group and In the HRZE treatment group, “Time” indicates the time before and after the patient was treated with antibiotics, and “:” indicates the interaction item. C. Principal Coordinate analysis (PCoA) based on the Bray-Curtis distance algorithm shows the differences in the intestinal microbial community structure between subjects before and after 14 days of treatment with HRZE and NTZ. The gray line connects the baseline and the 14th day treatment paired samples. PCoA1 clearly distinguishes the NTZ treatment sample from the baseline or HRZE treatment sample. D. Use the Inverse Simpson diversity index to analyze the diversity of the intestinal microbiota a of patients. Among them, HRZE group: n=16; NTZ group: n=19. Use a linear mixed-effects model to determine the importance of differential treatment to diversity. Using the linear mixed effects model (Diversity~1+Sex+Age+Treatment: Time +1|ID), “:” represents the interaction term, and HRZE is used as the reference level. At baseline, there were no significant differences between the two treatments. After 14 days of treatment, both groups showed a significant decrease in a diversity.
2 In patients treated with NTZ, the changes in the composition of the gut microbiota caused by antibiotics are more obvious
In order to identify the groups significantly affected by the two interventions, the researchers performed linear mixed effects modeling (ASVi (counts)) based on Limma/voom on the abundance of each sequencing variable identified by dada2 (ASV) (ASVi (counts)~Sex+ Batch+Group+1 | ID). In addition to determining the effects of treatments (pretreatment, HRZE, NTZ), the model can also quantify the dependent effects between gender and sequencing batches. The researchers used the 1| ID random effect to control the baseline differences between different individuals, and used Benjamini-Hochberg FDR corrected p-values to identify ASVs that were significantly affected by treatment.
The above analysis shows that the effect of HRZE treatment on the gastrointestinal microbes of patients includes the exhaustion of 82 ASVs, most of which belong to Clostridiales (FDR<0.05). It is worth noting that most of these ASVs (such as Blautia spp., Butyrivibrio spp., Clostridium spp., Eubacterium spp., Faecalibacterium spp., Gracilibacter spp., Oscillibacter spp., Roseburia spp., Ruminococcus sppacter spp., .) are involved in host health-related functions, such as SCFA production or bile acid conversion (Figure 3A, B). NTZ treatment has a more substantial interference on the intestinal flora of patients, with a total of 387 ASVs depleted and 16 ASVs enhanced. NTZ treatment caused a significant reduction of Clostridiales in the intestine, which included all ASVs except the one cleared by HRZE treatment, but also many other Firmicutes (FDR <0.05). In addition, NTZ caused the expansion of pathogenic bacteria including K. pneumoniae, E. coli, C. freundii, S. alactolyticus and E. faecium (FDR <0.01) (Figure 3A, B). Bacilli and Enterobacteriaceae can be screened out through antibiotic treatment, because higher concentrations of antibiotics can induce redox potential, including increased redox in intestinal epithelial cells.
In summary, the above results show that although NTZ treatment has no significant effect on the Mtb load in patients, it has more significant disturbances to the patient’s intestinal microbiota than HRZE treatment, including the exhaustion of a large amount of Clostridia, and screening a class Oxygen-resistant pathogens associated with known diseases.
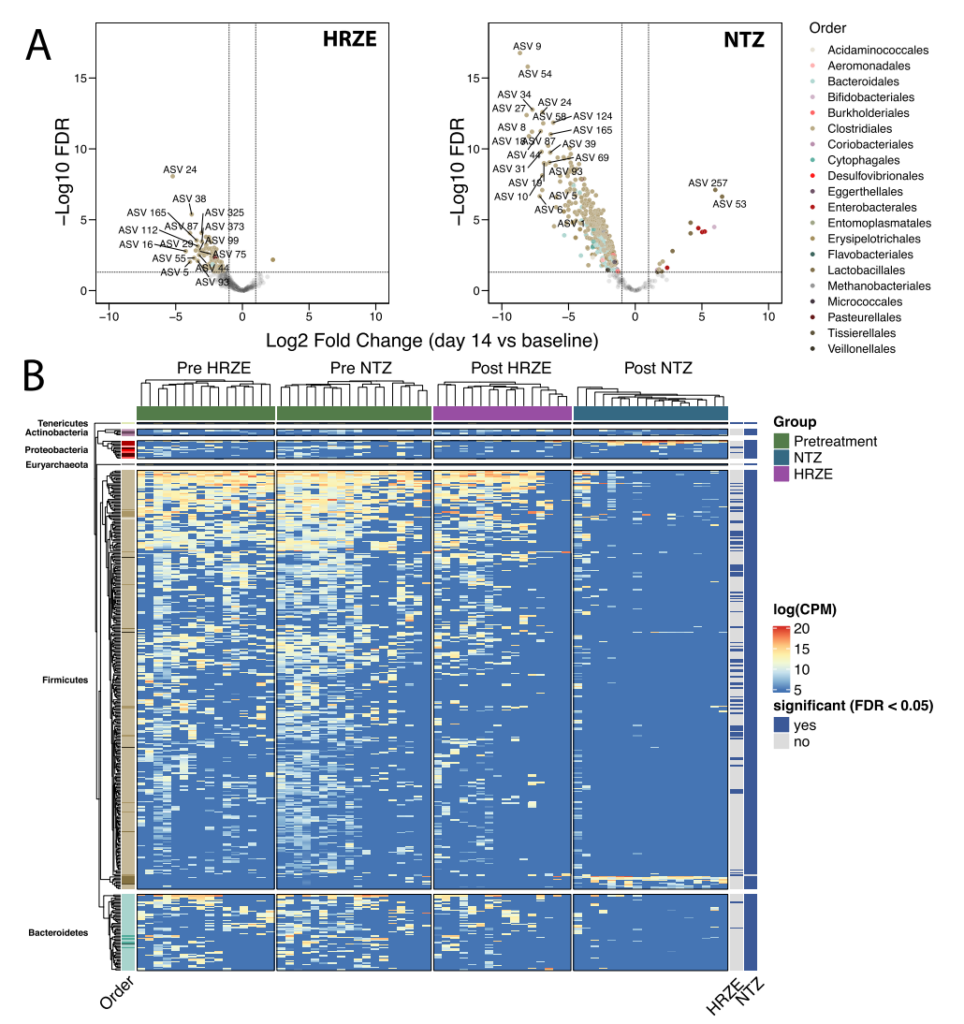
Figure 3 NTZ and HRZE treatments triggered changes in the gut microbiome of subjects. A. Volcano chart shows the difference between HRZE and NTZ treatment (day 14) and pretreatment (baseline) based on ASV levels. The color of each ASV is arranged in the order of the phylogeny of the strain. A single linear mixed-effect model (ASVi(counts)~Sex+Batch+Group+1|ID) of each ASV was fitted to determine the differences due to processing factors, taking into account sequencing batches and gender. B. Unsupervised hierarchical cluster analysis of the abundance of 404 ASVs significantly affected by HRZE or NTZ intervention (FDR <0.05). The annotations on the right (HRZE and NTZ) indicate whether each ASV is significantly interfered by treatment. The P value on the y-axis is adjusted according to the Benjamini-Hochberg algorithm to calculate the FDR.
3 HRZE and NTZ specifically affect the expression of host peripheral genes
Because the above results highlight the different effects of the two therapies on the treatment of tuberculosis and the composition of the gastrointestinal tract, the researchers realized that this provides a unique opportunity to speculate on the composition of the intestinal microbiota, the bacterial load and the peripheral Possible associations between gene expression. Because subjects have a certain degree of randomness before being assigned to the above two treatment options, the researchers used Limma/Voom’s linear mixed effects model to model the abundance of each host transcript (Genei (Counts) ~Sex+Batch+Group+1|ID) analysis.
The study used gene set enrichment analysis (GSEA) on limma/voom expression data compared with baseline and NTZ or HRZE treatment to determine the functional pattern of overall transcript abundance changes caused by treatment. The study used GSEA to assess the degree of enrichment of the MiSigDB Hallmark pathway, with the goal of providing a broad overview of the biological pathways that may be expressed.
In subjects treated with HRZE, the researchers observed a significant reduction in inflammatory response on day 14 (FDR <0.05), including IFNa response, IFNg response, and NFκB-mediated TNFa pathway and IL6 JAK STAT3 pathway, the above response It is related to the systemic immunological effects of antibiotics that reduce the level of bacterial pathogens (Figure 4A). In contrast, NTZ treatment showed the opposite effect. HRZE treatment down-regulated inflammation-related signaling pathways, including TNFa signaling pathway, IFNg signaling pathway, and type I interferon signaling pathway, and they were significantly enriched after NTZ treatment on the 14th day (Figure 4A).
NTZ treatment also up-regulated several other pathways, such as hypoxia, apoptosis and reactive oxygen species (ROS), which are considered to be markers of immune disorders. Since the study found that NTZ treatment can change the composition of the patient’s intestinal microbiome, while keeping the Mtb bacterial load in sputum (TTP) basically unchanged, the researchers speculate that the above effects caused by NTZ may be part of the change in microbial composition Features.
In order to further understand the impact of these two drugs on gene characteristics, the researchers first concentrated on analyzing a set of published transcriptional markers of active tuberculosis. These markers came from multiple population cohorts and crossed sequencing platforms (microarray and RNAseq). ). After analysis, it is found that the above-mentioned markers are significantly different between LTBI, active tuberculosis and healthy control individuals.
In this study, the researchers detected 363 of 373 transcripts in pretreated active tuberculosis subjects. After two weeks of HRZE or NTZ treatment, the researchers defined three main types of changes in these transcripts: (1) Renormalization (the transcripts that changed before and after HRZE/NTZ treatment, compared with the active TB and control groups reported in the previous literature). The multiples of difference between LTBI have the same trend); (2) unchanged (no change in transcript expression before and after HRZE/NTZ treatment); (3) deterioration (changes in multiples of differential genes before and after HRZE/NTZ treatment are similar to those reported in the literature The fold change relationship between active TB and control/LTBI is opposite.
Among them, 157 active TB genes are significantly affected by HRZE treatment (FDR<0.05) (Figure 4B). Among the 157 affected genes, 144 (92%) gradually returned to normal as the treatment progressed (that is, active TB showed the same fold change trend compared with the control group), while 13 (8%) got worse (that is, The opposite direction of the fold change). On the other hand, the study also found that only 4 of these tuberculosis-related inflammation genes were affected by NTZ treatment, and they all belonged to the acute attack stage (Figure 4C).
Because NTZ treatment changes the gut microbial composition of the patient without affecting the remission rate of Mtb disease, the researchers speculate that there may be other host-related transcript subgroups that are related to the dependent immunity of gut microbes, which may have an effect on the above-mentioned microbial structure The changes are responsive. In order to test the above hypothesis, while taking into account the established link between microbial disorders and auto-inflammatory conditions, such as inflammatory bowel disease, the study selected a recently published set of 880 genes.
Compared with asymptomatic controls, the above-mentioned genes The expression is significantly different in colon biopsy tissues of IBD patients. Among these 880 genes, 364 were detected in the data of this study. The researchers defined renormalization as a gene that has the same fold change as the control/active IBD through antibacterial treatment. Although compared with NTZ treatment, HRZE intervention has a more limited impact on the gut microbiome of patients, but the study still found that HRZE treatment resulted in 117 gene changes (FDR <0.05) (Figure 4D), of which NTZ treatment was associated with 55 The change of each gene is related (FDR <0.05) (Figure 4E). The above results indicate that HRZE and NTZ treatments may cause microbial-related peripheral inflammation changes in patients.
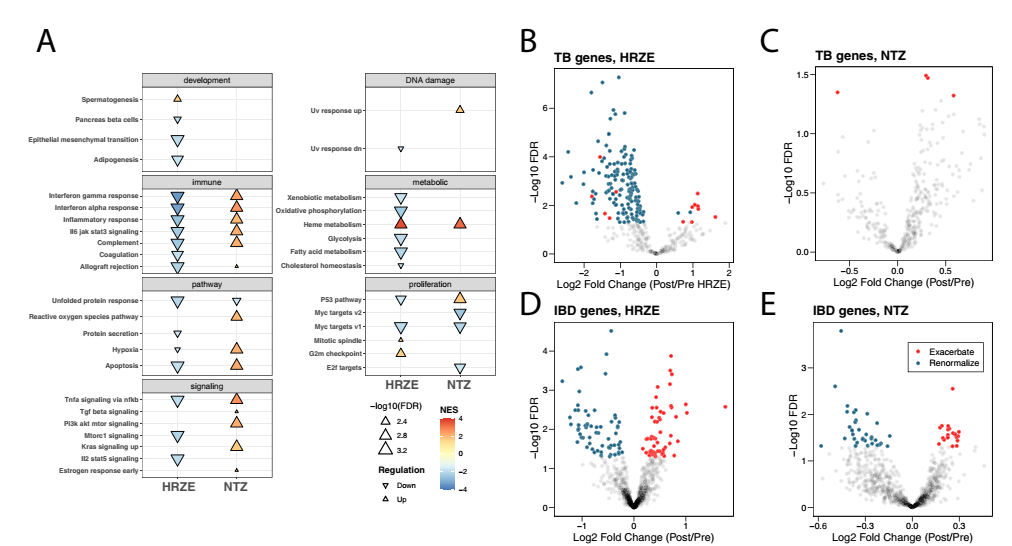
Figure 4 Hallmark pathway gene set enrichment analysis and gene expression comparison of HRZE and NTZ treatment groups. Hallmark gene pathway changes are associated with HRZE (A) or NTZ (B) treatment for 2 weeks. Compared with baseline, positive means that the pathway is overexpressed (up) at 2 weeks of treatment, and negative means that pathway expression is down-regulated (down) at 2 weeks of treatment. The size of the arrow indicates the level of significance. Peripheral blood transcripts related to tuberculosis treatment emphasize the difference in HRZE (B) or NTZ (C) gene expression from baseline after treatment. HRZE treatment re-normalized the expression of 144 effective TB inflammation-related transcripts, that is, close to a healthy state, and only up-regulated the expression of 13 inflammation transcripts. However, NTZ treatment up-regulated 4 inflammatory transcripts. The significance of each treatment affecting the TB-related peripheral blood transcription level is defined as in the limma/voom model, for the variable Group, the p value of its FDR is less than 0.05. HRZE treatment (D) and NTZ (E) lead to renormalization (HRZE 66, NTZ 34) or exacerbation (HRZE 55, NTZ 21) of different genes. The significance of IBD-related peripheral blood transcripts affected by different treatments is defined as in the limma/voom model, for the variable Group, the p value of its FDR is less than 0.05. The P value on the y-axis is adjusted according to the Benjamini-Hochberg algorithm to calculate the FDR.
4 Longitudinal observation and analysis of intestinal microbes and peripheral inflammation of patients in the HRZE treatment cohort
In order to verify the observation results after two weeks of HRZE treatment and determine the possible impact with the extension of treatment time, the study recruited a longitudinal treatment cohort (20 persons) to evaluate the remission of the disease during the 6-month period of HRZE treatment, and Regularly sampling patient saliva samples for TTP testing, stool samples for microbiome analysis, and peripheral blood samples for transcriptome sequencing (Figure 5A).
All participants received standard HRZE treatment (isoniazid 300 mg per day, rifampicin 600 mg per day, pyrazinamide 25 mg/kg per day, and ethambutol 15 mg/kg per day). The bacterial load (TTP) of Mycobacterium tuberculosis in saliva samples was detected on the 7th day, 14th day, 1 month, 2 months, and 6 months at the baseline of treatment. At the same time, the patient’s bacterial load (TTP) was also collected at each time point. Stool samples were used for microbiome sequencing analysis, and patients’ whole blood was collected for transcriptomics analysis at baseline, 14 days and 2 months.
The above results are similar to those observed in the HRZE intervention group in HRZE/NTZ. After two weeks of treatment, TTP was significantly higher than baseline (p <0.05). At the same time, the TTP after two months of intervention was also significantly higher than that at two weeks. This result shows that the antibiotic treatment has a bactericidal effect on Mycobacterium tuberculosis (p <0.05) (Figure 5B). Longitudinal analysis of the changes in the microbiome structure of the patient showed that the diversity of the gut microbiota of the patient decreased significantly after 7 days of treatment, and increased after 1 month of treatment, but was still significantly lower than the baseline level at 6 months of follow-up (p <0.05) (Figure 5C).
The Limma/Voom difference analysis was performed using the same method as above to determine the effect of the treatment method and time on the patient’s gut microbiome and peripheral transcriptome. As the researchers’ previous results are consistent, it was found that HRZE treatment can significantly reduce the abundance of Clostridiales in the intestine after one week of treatment.
Compared with baseline, most of the above-mentioned Clostridiales ASVs were even at the 6-month follow-up time point. Still significantly reduced, (Figure 5D). Overall, compared with baseline, on day 7, 19 ASVs were depleted and 3 ASVs increased; on day 14, 61 ASVs were depleted and 2 ASVs increased; and at one month, compared with baseline In comparison, 83 ASVs were exhausted and no new ones were added. Therefore, in the first month of treatment, compared to the baseline sample, the diversity of the subject’s gut microbiome was significantly reduced.
However, in the later stages of the treatment process, the study found different trends. Relative to baseline, compared with day 0 of two months of HRZE treatment, the study found that 11 ASVs had a reduced abundance and 53 ASVs had an increased abundance. At the 6th month of HRZE treatment, compared with day 0, the study found that 3 ASVs were significantly reduced, while 327 ASVs increased (Figure 5D). At 2 and 6 months, the rise of specific ASVs is relatively heterogeneous among different patients. In addition, some subjects with no Clostridiales ASVs detected on day 0 showed enrichment at the next two time points.
Overall, the longitudinal analysis shows that after one month of HRZE treatment, most ASVs that may be affected by the treatment will be exhausted, but will gradually recover after 2 to 6 months of treatment. In addition, the results also indicate that ASVs related to phylogeny that were not detected on day 0 may replace certain flora.
Compared with baseline, at two weeks (day 14) and two months of treatment, the study found significant changes in the subjects’ peripheral inflammation. The down-regulation of common inflammatory pathways was found in the HRZE/NTZ trial cohort (Figure 5E). Interestingly, comparing the data on day 56 with baseline or day 14, the study found that inflammation genes were further reduced, which may be due to a further increase in TTP on day 56 (Figure 5F-K).
Mathematical modeling with multiple omics constraints can distinguish the contribution of gastrointestinal microorganisms and Mycobacterium tuberculosis to the characteristics of peripheral inflammation. Next, the researchers tried to determine the relative contribution of the dynamic changes of gastrointestinal microbes and Mtb in predicting changes in the trend of peripheral inflammation genes.
As a basic reflection of the inflammatory state of tuberculosis, the study concluded that the mitigation of this state may be affected by the combination of upstream factors such as pathogen clearance and microbiome changes. The researchers hypothesized that these relationships could be modeled by using individual variability through treatment with HRZE and using data from the NTZ group in the HRZE/NTZ trial cohort. This is because the NTZ group can actually be used as an effective experimental control to detect changes in gastrointestinal microbiota and gene expression without changes in Mtb.
Specifically, the goal of the study is to determine the specific microbial populations in which changes in flora abundance are associated with significant changes in inflammatory pathways in the three groups of antibiotic-treated patients. To train the above model, the researchers used all the results in the HRZE/NTZ trial cohort and the longitudinal observation cohort, a total of 34 paired samples (Figure 1). Simultaneously use all data from two longitudinal cohorts (HRZE/NTZ trial and HRZE longitudinal observation cohort) to fit each inflammatory pathway to discover patterns that are common in multiple data sets.
Each model is 80%-20% The data is partitioned for training and verification. This method is suitable for common types of multi-omics data sets in clinical research. Importantly, compared with traditional multiple linear regression analysis, RFR modeling has obvious advantages, because it has nothing to do with model structure (for example, non-parametric regression), does not need to meet the general assumptions required by classical regression analysis, and can be executed Set ranking feature selection.
What’s important is that although the interpretation of RFR is less straightforward than traditional regression analysis (for example, there is no regression coefficient itself), its downstream analysis includes Permutated importance and Accumulated local effects (ALE). ) Can be used to evaluate the importance of predictive factors (such as TTP, microbiome composition, etc.) and their impact on dependent variables (such as markers of host peripheral inflammation).
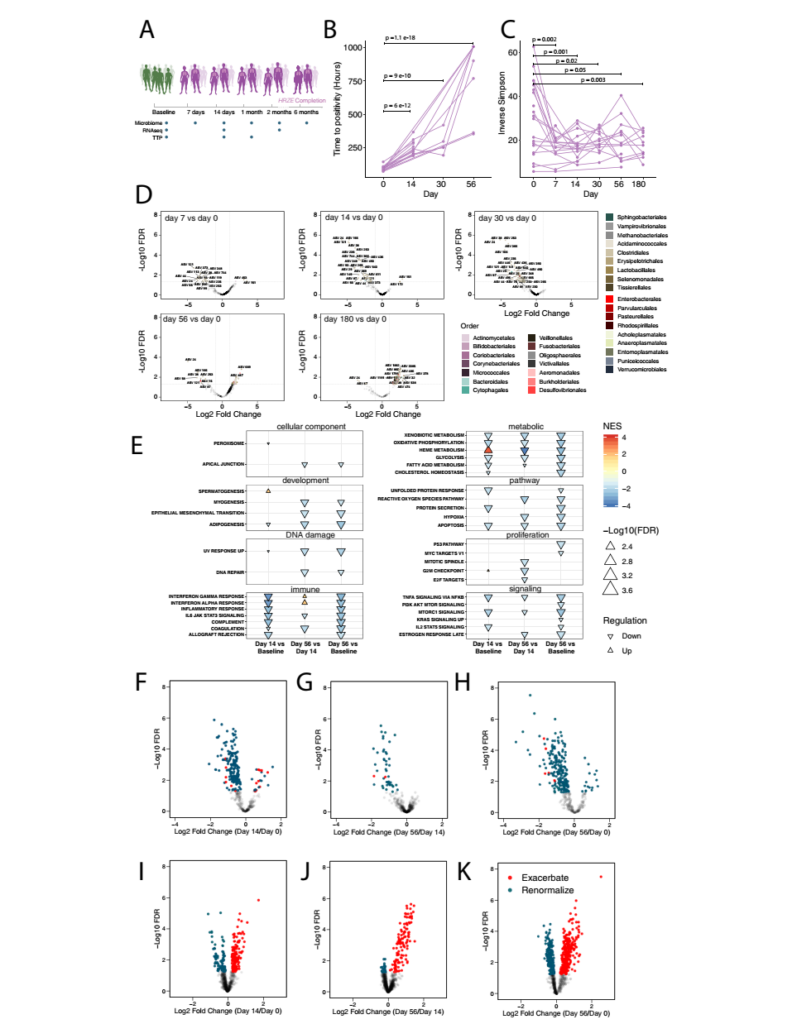
Figure 5 HRZE treatment causes longitudinal changes in the composition of the gut microbes and the expression of peripheral inflammation-related genes. A. Schematic diagram of the study cohort. B. The time to be positive at baseline (19 biological independent samples), day 14 (14 biological independent samples), 1 month (5 biological independent samples) and 2 months (9 biological independent samples). In order to determine the statistical significance of the difference in TTP at different time points, the study fitted a linear mixed effects model (TTP ~ Sex+Age +Time+ 1|ID). The investigator checked the p value associated with the time variable to determine the significant difference in TTP (p value <0.05). C. Calculate the baseline (20 biological independent samples), day 7 (7 biological independent samples), 14th day (19 biological independent samples), 1 month (13 biological independent samples) for each subject , 2 months (13 independent biological samples) and 6 months (13 independent biological samples) of the microbial community diversity, using the Inverse Simpson index to calculate the a diversity of the microbiome. D. The volcano map shows the difference between the microbial community composition and the fold change on the 7th, 14th, 30th, 56th and 180th days. E. The normalized enrichment score calculated by Hallmark Pathway between Day 14 and Baseline, Day 56 and Day 14, and Day 56 and Baseline. The F-H volcano graph shows the difference in TB transcription levels between the 14th day and the baseline, the 56th day and the 14th day, and the 56th day and the baseline after treatment. The I-K volcano plot shows the difference in transcription levels between IBD cases and the control group detected between day 14 and baseline, day 56 and day 14, and day 56 and baseline in the study of Palmer et al.
When plotting the average slope of the ALE curve for predictors with significant ranking importance values (p <0.05), the study found an increase in TTP (decrease in Mtb in sputum) and an increase in the abundance of Clostridia ASVs, especially Clostridia IV And XVIa, which have been shown to induce anti-inflammatory responses through the production of SCFA E rectale, F. prausnitzii, G. formicilis, E hallii, O ruminantium, D. formicerans, S. variable, B. faecis, and B. obeum . The abundance of the above-mentioned flora is related to the reduction of peripheral pro-inflammatory response, including INFγ, INFa, inflammatory response and IL6 JAK STAT3 signal transduction (Figure 6). In contrast, the increased abundance of E. coli and E. faecium is associated with increased inflammation (Figure 6), indicating that the overgrowth of these species in the gastrointestinal tract is a sign of microbial disorders and inflammation, and is usually associated with poor clinical outcomes.
In summary, the data of the study and related calculation analysis show that the changes in inflammatory gene expression accompanying tuberculosis treatment are related to the antibacterial activity of drugs that lead to pathogen clearance and changes in the composition of microorganisms induced by antibiotics. Based on the results of this modeling, the researchers proposed two modes of microbe-related inflammation. The first is that the depletion of Clostridia (especially Clostridia IV and XVIa) exacerbates the TB-related inflammatory response, which is significantly different between the HRZE and NTZ treatment groups. In addition, the increase of pathogens that are only related to NTZ treatment, such as E. faecium, S. alactolyticus and E. coli, may also exacerbate the up-regulation of inflammatory pathways in individuals. It is important that the model of this study clearly illustrates the abundance of all ASVs affected by the treatment (ie, the presence and abundance of all bacteria are controlled by using the microbial species in the model as a predictor). Therefore, the correlation between the abundance of the aforementioned pathogens and the intensification of host inflammation-related reactions indicates that the established correlation between microbiome changes and host immune response may not only reflect the dynamic changes of Clostridia.
Based on this model, studies have also hypothesized that the remission of tuberculosis may be related to the preservation of clostridia, and their depletion and the enhancement of enterobacteriaceae bacteria or bacillus associated with pathogenic diseases may slow the ablation and even support the exacerbation of inflammation.
Based on this model, the researchers hypothesized that the remission of tuberculosis may be related to the increase in the abundance of Clostridia (especially Clostridia IV and XVIa), and its depletion in the intestines and subsequent increases in Enterobacteriaceae and Bacilli pathobionts associated with the imbalance of the flora It may prevent the disease from alleviating or even exacerbate the deterioration of inflammation.
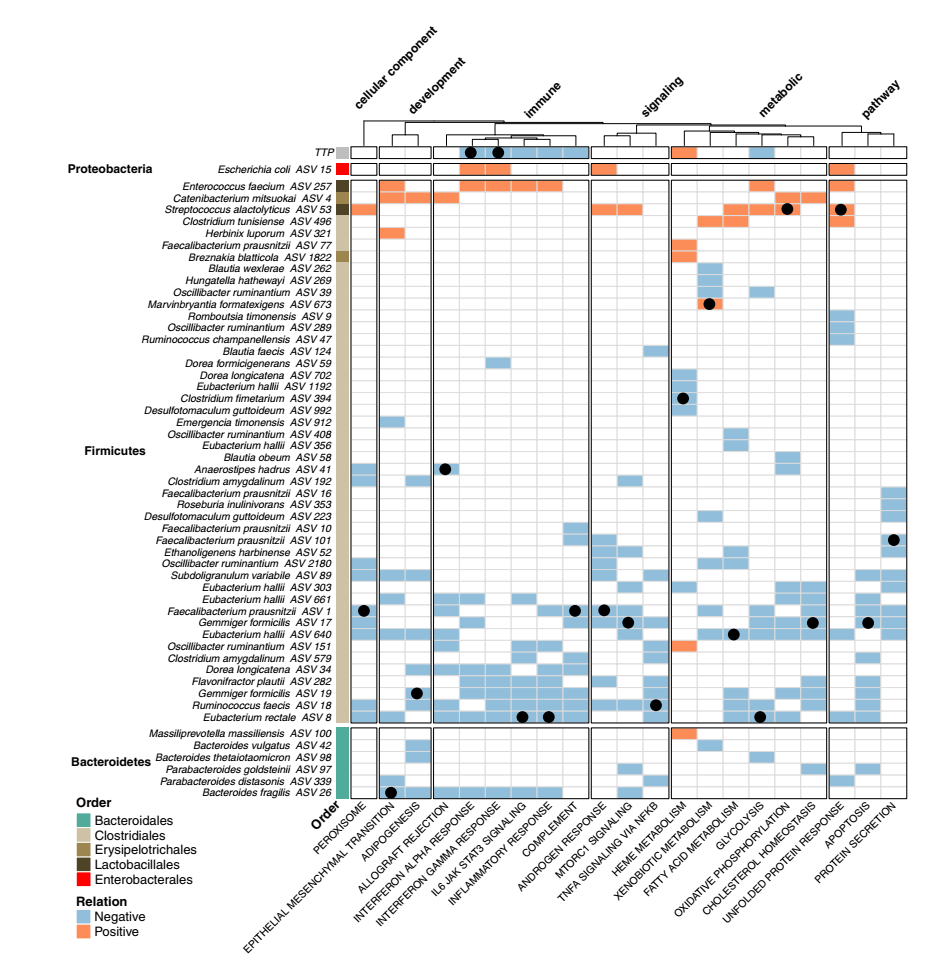
Figure 6 uses random forest regression to analyze the relationship between host immune-related peripheral blood gene characteristics and gut microbial composition and TTP changes. The heat map shows the changing trend of the ALE curve. Blue/orange indicates features that are significantly related to changes in specific inflammatory pathways, blue indicates negative correlation, and orange indicates positive correlation. For each immune pathway, the p-value of Benjamini-Hochberg FDR adjusted from the substitution importance analysis is less than 0.05, and it is considered that there is a significant association between the pathway and ASV. The black dots are used to identify the most important predictors of the modeling identification for each specific host path (ie, the predictor that will cause the largest increase in the mean square error between the model prediction and the result if it is missing). This analysis shows that the decrease in TB-related load and the increase in the abundance of Clostridia IV and XIVa related to host health can be used to predict the reduction of host inflammatory response. On the contrary, an increase in the abundance of aerobic bacteria (including Enterococcus, Streptococcus and E.coli) in the patient’s intestine indicates an increase in the host’s inflammatory response.
5 Verify the relationship between the gut microbiome and peripheral gene expression in healthy controls in the cohort
Researchers performed machine learning modeling on data from two longitudinal treatment cohorts and showed that specific gut microbiota groups are related to immune-related gene characteristics in the host’s peripheral blood. Specifically, the high abundance of Clostridia is negatively correlated with inflammation (such as interferon a, interferon g, IL6/JAK/STAT3, inflammatory response gene signals), and the high abundance of common oxygen-resistant pathogens is associated with the deterioration of the above-mentioned inflammation related. In order to assess the universality of the above findings, the researchers speculated that even in healthy individuals, the different levels of bacterial colonization are related to the different levels of host immune-related peripheral blood genes.
In order to test this hypothesis and finally verify it from the model, the researchers analyzed a set of population data from two healthy control cohorts. A subset of these data has been reported in the published work of the research team, namely a cross-section of TB-negative healthy family contacts (FC) and healthy non-contacts (Community Controls, CC) from the same community in Haiti the study. For these two cohorts, the study included a total of 55 healthy controls, including 9 FCs and 36 CCs. The researchers collected microbiome 16S rRNA sequencing data and peripheral blood transcriptome data.
The researchers first tried to verify the above results, that is, in the verification control cohort of FC and CC, the peripheral blood transcription pattern of subjects returned to normal after HRZE treatment, and deteriorated after NTZ treatment (this is through our comparison with the published control/ The comparison of the active tuberculosis gene signature data set is confirmed) is still valid (cohort 3). In order to link the transcript abundance with the enrichment of immune pathways, the researchers used the R language GSVA software package to perform a single-sample gene set enrichment analysis.
The researchers first performed an unsupervised cluster analysis of the NES scores of the pathway samples of all samples in this study, and found that individuals from different cohorts have wide differences in different biological pathways (Figure 7A). Then, the researchers used linear mixed-effect models for the HRZE/NTZ trial and the long-term observation cohort, respectively, to compare the Euclidean distance between each pair of FC/CC and the sample before or during treatment with time, treatment, and patient ID.
Perform regression analysis. It was found that in the HRZE/NTZ trial and the long-term observation cohort, compared with before treatment (day 0), HRZE treatment significantly shortened the distance to FC/CC samples (the p value of HRZE in the HRZE/NTZ trial < 1e-20, longitudinal observational HRZE treatment cohort <1e-10, longitudinal observational HRZE treatment cohort, p value <0.05). On the contrary, in the HRZE/NTZ trial, compared with the p value before treatment, NTZ intervention increased the distance to FC/CC. This result once again verified the increase in the inflammatory response in patients induced by NTZ.
The study performed RFR modeling analysis on the microbiome data of each pathway, and summarized the results in Figure 7B. Surprisingly, researchers found that a large number of Firmicutes, especially Clostridia, are associated with many characteristic molecular pathways. More interestingly, the study found that Clostridia related to host health, including F. prausnitzii, Rumonoccocus spp. and C. catus, have higher abundance of ASVs, which is related to the reduction of pro-inflammatory pathways including INFA and INFg. This independent analysis under steady-state conditions is consistent with the results of the study’s longitudinal observational modeling analysis in the HRZE/NTZ cohort and the HRZE treatment cohort (Figure 6). The above results were carried out in a cohort of 55 volunteers from the Haitian community, thus further explaining that the relative abundance of specific gastrointestinal microbiota is related to the expression of peripheral genes related to the host inflammatory response, and these specific microbial groups Significantly affected in the treatment of tuberculosis.
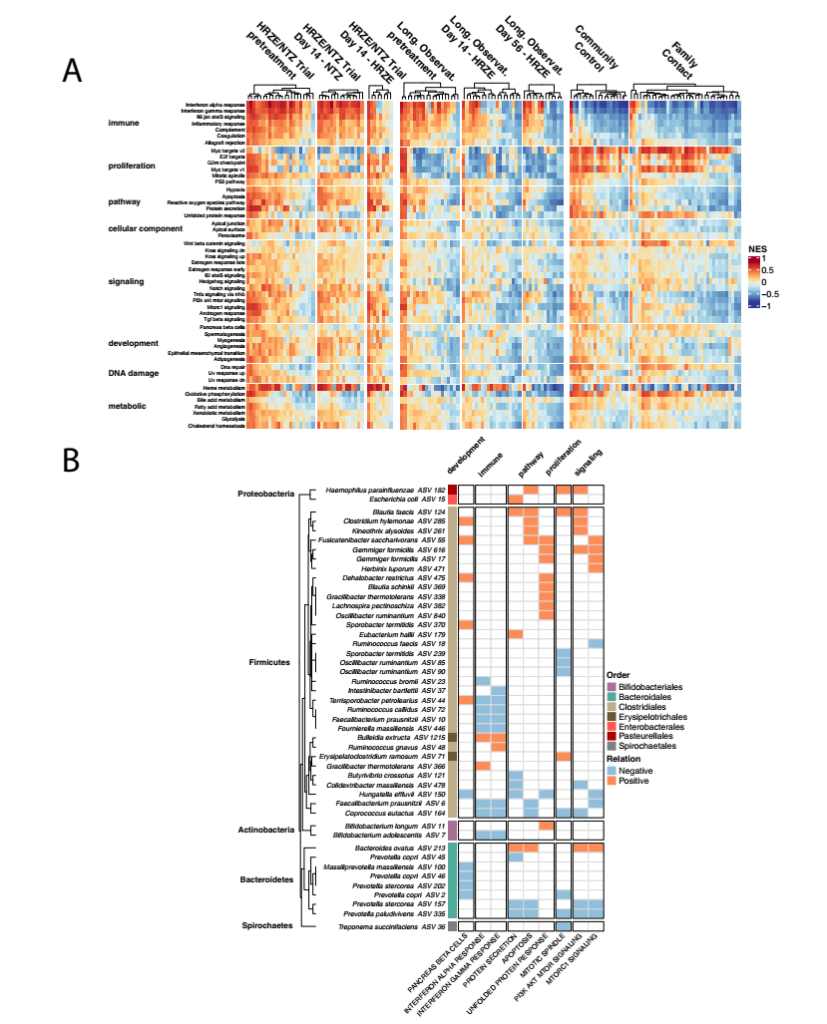
Figure 7 Analysis of the microbiome and blood peripheral gene expression of an independent healthy control population confirmed the association between specific gut microbes and host peripheral gene expression. A. For all cohorts in this study, NES scores based on the 50 Hallmark pathways of MiSigDB for each sample. Use DSEeq’s variance stabilization transformation count to calculate the NES score, calculate it with the GSVA software package in R, and scale it in all samples (Z score). B. Random forest regression analysis that associates specific microbial groups with Hallmark pathways. Only the paths identified in the RFR model are shown in the figure. The “Relation” calculated by taking the first derivative of the ALE diagram of each relationship. If the path is positively related to a specific ASV, then the “Relation” is positive; if the path is negatively related to a specific ASV, then the ” Relation” is negative.
Discussion:
Since the advent of the microbiome high-throughput sequencing technology, researchers have clearly realized that the use of antibiotics is one of the most effective means to identify microorganisms in the body, and its effects are acute and persistent. Specific microorganisms can have specific effects on the host’s immune effect, including affecting the abundance and function of immune cell subpopulations. A large amount of mouse-based research data has proved the influence of antibiotics on the composition and function of microorganisms in the body, as well as the subsequent influence of these microorganisms on host immune cells.
However, the extent to which antibiotic-induced microbiota disturbance can significantly change the results of anti-infective treatments, or the relationship between changes in the composition of the human intestinal microbiota and peripheral gene expression is still unknown. It is generally believed that the role of antibiotics in clearing infections is to directly kill pathogens and regulate immune responses through the microbiota.
The killing of pathogens by antibiotics may also be partially offset by harmful immune responses caused by microbiota disturbances. The above-mentioned dynamic process may be particularly related to the treatment of chronic infections such as tuberculosis. In this disease state, the duration of antibiotic treatment is prolonged, and the progress of the disease reflects the burden of pathogens in the body and the result of chronic symptom-related inflammatory mediators that lead to tissue destruction.
Tuberculosis patients who are sensitive to antibiotics usually need to use antibiotics for six months, mainly based on mycobacteria-specific drug treatment. In this study, the researchers reported the effects of HRZE treatment on the early and late gut microbiome of patients with active tuberculosis, and demonstrated that after only two treatments, observations in a cross-sectional study of tuberculosis patients The same trend of change (compare cured patients and LTBI individuals).
After several weeks of treatment, the study found that under the treatment conditions of HRZE, the abundance of Clostridia in the patient’s intestine was significantly affected, and this finding was also confirmed in mouse experiments. In view of the specificity of mycobacteria targeted by the treatment of tuberculosis drugs and the interaction of small molecules that affect the combination of microbes, it is difficult to predict that HRZE therapy will mainly target Clostridia in the Firmicutes phylum, while the Actinomycetes to which Mtb belongs is not affected. influences. Experimental results in mice indicate that this anti-clostridium effect is mainly driven by rifampicin/PZA treatment. Clostridia is an immunologically active component in the gut microbiota, which can produce a variety of metabolites, such as short-chain fatty acids and other compounds.
Research based on NTZ/HRZE enables this study to analyze the relative contribution of pathogen removal and microbiome disturbance to disease treatment. Because the standard treatment both reduces the Mtb bacterial compliance and disturbs the microbiome in the body, NTZ has no significant effect on the average load of Mtb, but it does interfere with the microbial composition in the body in a way that overlaps with HRZE. At the same time, the above findings were expanded in the longitudinal cohort, which tracked the changes in patients with active tuberculosis one week, two weeks, one month, two months and six months after treatment, confirming that the use of antibiotics affects the body The disturbance of the microbiome will have a systemic effect on the expression of genes in the host’s periphery.
Furthermore, the study speculates that the huge difference in patient response to tuberculosis treatment may be due to the partial effect of antibiotics on the heterogeneity of the microbiota.
In order to verify the inferred potential relationship between microbiota and host inflammation, the researchers conducted microbiome and blood transcriptome analysis from an independent Haitian healthy population cohort. It is worth noting that although the peripheral level of the inflammatory pathway is reduced compared with patients with active tuberculosis, the study once again found that the increased abundance of Clostridium IV and XIVA is related to the decreased expression of the pro-inflammatory pathway. The above results support the conclusion of this study that whether in the disease state of tuberculosis or under steady-state conditions, the composition of the microbiome determines the systemic inflammatory state.
Further, considering the challenge of using paired samples to explain the relationship between the composition of the microbiome and the expression of surrounding genes, the samples were randomly allocated to drug combinations that have significant differences in the systemic effects of the body, and an appropriate algorithm was attempted to carry out this Class analysis. For single-omics microbiome and RNA seq data, the study chose to use limma/voom to simulate the changes in these data because of its ability to model multiple effects and the subject’s baseline value as Random effects. For multi-omics integration data, the study used random forest regression.
The above analysis results highlight the usefulness of these models in two ways: (1) by providing evidence that supports or opposes specific hypotheses about the clinically significant relationship between many potentially relevant parameters, and (2) by providing evidence of these models The hypothesis-generating relationships between multiple components (ie features) can be further tested in mice, validation cohorts, or other model systems.
One of the main goals of the research is to infer the relationship between human gut microbiota, pathogen load and host inflammation by using fast and non-invasive measurement methods (for example, fecal DNA sequencing, saliva bacterial load and blood sample transcriptome sequencing) contact. The underlying theoretical basis is that the analysis of these associations can guide follow-up mechanistic research in vivo and in vitro, not only for verification, but also for establishing intervention strategies targeting the microbiome, thereby improving inflammation remission during tuberculosis treatment .
The above analysis is based on the predictive variables that TTP is sufficient to predict the impact of tuberculosis on the systemic immune response. In addition, it is also necessary to consider that changes in the gut microbiome may not only affect the systemic inflammatory response, but may also affect the type and number of immune cells retained in the lungs. In addition, any correlation results obtained in this study may not fully explain the underlying variables or confounding factors of the relationship between the body’s intestinal microbe-inflammatory response-pathogen.
At the same time, although the above analysis was carried out in an independent cohort, the correlation analysis of the peripheral gene expression of the identified microorganisms is also applicable to the healthy control cohort, and the verification of people without tuberculosis but with other diseases may be It is more likely to produce false positive results than healthy people. Therefore, future research work will focus on determining whether the above-identified association exists in a new independent tuberculosis treatment cohort.
This data shows that within the first 14 days of treatment for tuberculosis patients, the resolution of the inflammatory response in the patient’s body (through the analysis of peripheral blood transcriptomics) may be affected by both the decrease in the body’s Mtb load and the disturbance of the intestinal microbiota induced by antibiotics. The latter may directly affect systemic immune function changes. Among the pathways closely related to these two factors, one is the characteristic activation pathway of active tuberculosis: interferon gamma, type I interferon and tumor necrosis factor alpha.
More and more evidence shows that the results of active tuberculosis reflect the burden of pathogens in the body and the mixed effects of cytokines including IL-1 and IFNγ, and the latter aggravates the progression of the disease. The study showed that the disturbance of the gut microbiome that accompanies tuberculosis treatment is one of the predictive markers of the normalization of these same pathways during early treatment, indicating that changes in the gut microbiome can affect or predict the rate of remission of tuberculosis.
In the first two weeks of treatment, pathogen killing is the dominant factor, but in the later stage of treatment, when pathogen killing slows down, the inflammatory response-related microbial composition-dependent regulation may play an important role in the treatment process. In the healthy control cohort, the verification of the relationship between gut microbial composition and peripheral gene expression, especially the co-expression of the same pro-inflammatory and anti-inflammatory pathways mentioned above, indicates that these associations can be extended to other populations.
Whether the above-mentioned association is causal or as a biomarker of another state will be the frontier direction of this research in the future. The next research work will be devoted to applying the analysis tools and research design ideas proposed in this study to the later stage of tuberculosis treatment to check whether the changes in the intestinal microbiota during the treatment process are related to clinically relevant treatment results, as well as Clostridia’s Whether the abundance is related to the elimination rate of Mtb in the body or the regression of the inflammatory response accompanying active tuberculosis.
These studies provide references for related studies targeting intestinal microbes as predictors of tuberculosis treatment, and at the same time help to further analyze the heterogeneity of treatment results between individuals.
Expert Comments:
Tuberculosis (tuberculosis, TB) is an infectious disease caused by Mycobacterium tuberculosis (Mtb) infection. There is an interaction between the intestinal flora and tuberculosis, which is mainly produced by the host immune system.
After Mtb infection, the body can produce a series of complex immune responses, this process involves a variety of immune cells and cytokines (such as CD4 + T cells, macrophages, TNF-a, IFN-g, etc.), especially CD4 + T Cells, the differentiated Th1/Th2 cell subsets can regulate the direction of the anti-tuberculosis immune response.
In view of the fact that standard treatment for tuberculosis can cause significant changes in the gastrointestinal microbiota, including the exhaustion of many Clostridium species, the above changes may be related to the body’s systemic inflammatory response. However, because HRZE therapy can quickly eliminate Mycobacterium tuberculosis in the early stage of treatment.
Therefore, if there is no Mtb pathogen killing or microbiota disturbance, and it is not a control group with both, it is difficult to separate the immune effect of eliminating Mtb from the interference of the intestinal microbiota. Therefore, the study combined three independent clinical cohorts, and analyzed the abundance of Mycobacterium tuberculosis in saliva samples by 16SrDNA sequencing, peripheral blood transcriptome analysis, and saliva samples.
By comparing standard tuberculosis treatments with experimental therapies that do not reduce tuberculosis bacteria load, the study can independently study the influence of intestinal flora on tuberculosis treatment, and pointed out that the composition of specific flora is related to the decline of inflammatory indicators during tuberculosis recovery.
The above studies provide new research ideas for exploring the influence of intestinal symbiotic flora on the occurrence and development of infectious diseases.
(source:internet, reference only)
Disclaimer of medicaltrend.org



