The history and current status of KRAS target
- Why Lecanemab’s Adoption Faces an Uphill Battle in US?
- Yogurt and High LDL Cholesterol: Can You Still Enjoy It?
- WHO Releases Global Influenza Vaccine Market Study in 2024
- HIV Infections Linked to Unlicensed Spa’s Vampire Facial Treatments
- A Single US$2.15-Million Injection to Block 90% of Cancer Cell Formation
- WIV: Prevention of New Disease X and Investigation of the Origin of COVID-19
The history and current status of KRAS target
The history and current status of KRAS target. The full name of the KRAS gene is the gene Kirsten Rats Sarcoma viral Oncogene Homolog.
Background
RAS is the first human oncogene to be discovered.
At the end of the 19th century, the research of masters such as Pasteur made modern medicine a great success in the field of anti-infection. In 1876, a Russian successfully transplanted tumor tissue from one dog to another. In 1908, two Danish scientists discovered that extracts from hen leukemia cells could infect other birds with leukemia. These observations and studies have led people to believe that tumors are also infectious diseases caused by viruses.
In 1910, Peyton Rous of the Rockefeller Institute discovered the first tumor virus Roussarcomavirus (RSV, Roussarcoma virus) from a hen from Long Island, New York. Since then, a series of RNA and DNA viruses that cause animal tumors have been discovered one after another. The pathogenic mechanism has also been reasonably explained: these viruses (RNA viruses through reverse transcriptase) can integrate tumor genes into infected host cells, induce malignant transformation and maintain the infinite division of tumor cells. PeytonRous, who discovered RSV, and Howard Temin, who discovered RNA virus reverse transcriptase, won the Nobel Prize in Physiology and Medicine in 1966 and 1975, respectively. So far, people are convinced that tumors are diseases caused by viruses.
Until 1974, J. Michael Bishop of UCSF and his postdoc Harold Varmus accidentally discovered through DNA probes that the tumor gene src previously found in the RSV virus was also present in uninfected normal cells. It turns out that tumor genes have long existed in the host’s genome. In ancient times, viruses obtained these gene fragments from host cells and modified them, which means that the tumor genes carried by the virus actually came from our own. These discoveries opened the door to modern tumor biology based on genetic variation. Bishop and Varmus also won the third Nobel Prize in this field in 1989.
Harvey and Kirsten et al. respectively discovered the mouse tumor genes HRAS and KRAS carried by a retrovirus similar to RSV in the 1960s. In 1982, Weinberg and other laboratories also discovered HRAS in human bladder cancer cells T24/EJ, making RAS the first human tumor gene discovered.
After more than 50 years have passed, the RAS gene has been found to be one of the most common mutated genes in tumors. 30% of tumors carry RAS mutations. If the upstream and downstream mutations of RAS regulatory factors and signaling pathways are included, almost all tumors are covered. RAS mutations every year It is the king of tumor genes that caused more than one million deaths. This is also very curious. What magical function does RAS have that makes tumor cells so dependent?
KRAS gene and related pathways
1. The concept of KRAS gene
The full name of the KRAS gene is the gene Kirsten Rats Sarcoma viral Oncogene Homolog. The protein encoded by the KARS gene is a small GTPase (smallGTPase), which belongs to the RAS superprotein family.
2. Classification of KRAS genes
In the human genome, there are 2 KRAS genes. One is KRAS1, located on the short arm of chromosome 6, and the other is KRAS2, located on the short arm of chromosome 12. Among them, KRAS1 is a “pseudogene” that cannot be transcribed into RNA, so it has no function. KRAS2 is the “true gene”, which can be transcribed and translated into protein and has biological activity.
Note: Generally, the KRAS gene and protein studied in the company and literature reports refer to the “KRAS2” gene and its protein product.
The KRAS gene belongs to the RAS gene family. Among the RAS gene family, there are NRAS (neuroblastoma-RAS) and HRAS (Harvey-RAS). HRAS, KRAS and NRAS, located on chromosomes 11, 12 and 1, respectively, are involved in intracellular signal transmission.
There are 2 variants of the KRAS protein. These two variants are due to the fact that exon 4 has two different splicing methods during the splicing process of RNA, resulting in two kinds of mRNA, and then two kinds of proteins (variants) are translated. These two proteins are called “KRAS4A” and “KRAS4B” respectively. The expression level of KRAS4B is significantly higher than that of KRAS4A. In general, KRAS4B is 5 times that of KRAS4A.
3. The structure and location of KRAS protein
KRAS protein has 188 amino acids and its molecular weight is 21.6KD. Guanine nucleoside binding protein with GTPase enzyme activity.
The KRAS protein is located on the inner side of the cell membrane and is connected to the cell membrane through a Farnesyl modification group [1]. Farnesyl is modified by post-translational protein and added to KRAS protein [3] under the action of farnesyl transferase [2].
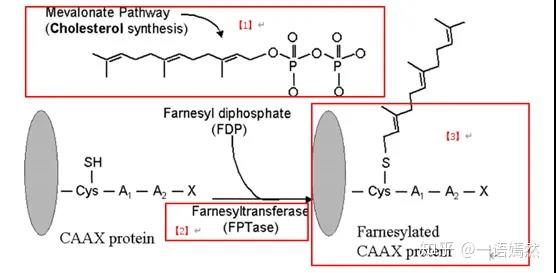
Source: Farnesyltransferaseinhibitorsinhematologicmalignancies:newhorizonsintherapy
4. KRAS related pathways
Upstream signaling pathway
In normal cells, receptor monomers such as EGFR, HER2, ErbB3 and ErbB4 on the cell membrane combine with extracellular ligands to form dimers, which are autophosphorylated and then phosphorylate downstream signal proteins. One of the signaling pathways can activate Grb2-Shc, then activate the SOS protein, and then activate the KRAS protein.
KRAS pathway
In the cell, the KRAS protein transitions between inactive and activated states. When KRAS binds to guanosine diphosphate (GDP), it is in an inactive state, and when it binds to guanosine triphosphate (GTP) At the time, it is in an active state and can activate downstream signal pathways.
Downstream signaling pathway
KRAS in most cells is in an inactive state. When it is activated, it can activate multiple downstream signaling pathways, including MAPK signaling pathway, PI3K signaling pathway, and Ral-GEFs signaling pathway. These signaling pathways play an important role in promoting cell survival, proliferation and cytokine release.
5. Regulators of KRAS activity
The transition of KRAS between inactivated and activated states is regulated by two types of factors.
One type is guanine nucleotide exchange factor (GEF), this type of protein catalyzes the binding of KRAS and GTP, thereby promoting the activation of KRAS, including SOS protein (genus GEFs/guanosine releasing factor/guanylate exchange factor).
The other is GTPase (GTPase) activating proteins (GAPs), which can promote the hydrolysis of GTP bound to KRAS into a GDP terminating active state, thereby inhibiting the activity of KRAS.
6. Adjustment process
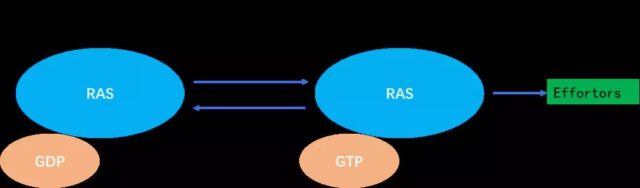
a) RAS is a binary molecular switch controlled by the GDP/GTP cycle:
When the KRAS protein is not activated, it is tightly bound to GDP and is in the “off” state; after receiving the signal, the guanylate exchange factor GEFs (SOS) are recruited The cell membrane binds to RAS and releases GDP. After KRAS is activated by the SOS protein, it binds to GTP.
b) The KRAS protein that binds to GTP has the activity of phosphokinase and represents the “on” state, which can further activate downstream proteins.
Note: When there is no stimulus signal, the switching of this off state is very slow.
c) Activated downstream signal proteins:
Raf protein family, PI3K protein, PLC-ε, RALGDS, TIAM1, RIN1 and other downstream proteins [1].
d) After the downstream signal pathway is opened, the downstream signal pathway is further activated [2], and cell functions such as cell proliferation and cell migration are initiated [3].
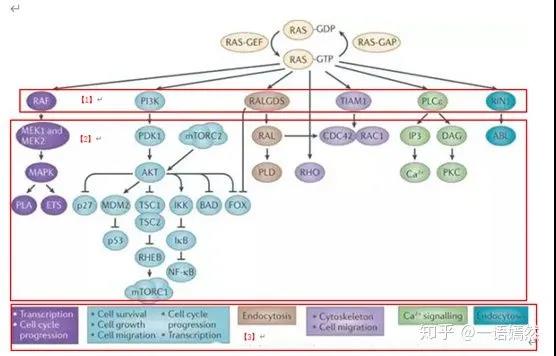
Image source: NatureReviews|Cancer
KRAS oncogene mutations
Because RAS is at the center of many important cell signaling networks, they are associated with many cancer markers. The RAS proteins are so named because they were first identified in the study of rat sarcoma caused by a strong carcinogenic virus. RAS is the most frequently mutated oncogene in human cancers. At present, mutation-induced activation of RAS protein has been found in about 1/5 of all human tumors.
KRAS is the most frequently occurring subtype in the RAS family. KRAS gene mutations account for 85% of the total RAS gene mutations (NRAS (12%) followed by HRAS (3%)). In human cancers, KRAS gene mutations occur in nearly 90% of pancreatic cancers, 30-40% of colon cancers, 17% of endometrial cancers, and 15-20% of lung cancers (mostly NSCLC). It can also occur in cancer types such as bile duct cancer, cervical cancer, bladder cancer, liver cancer, and breast cancer. That is to say, in the above-mentioned multiple cancers, there is a high proportion of KRAS gene mutations.
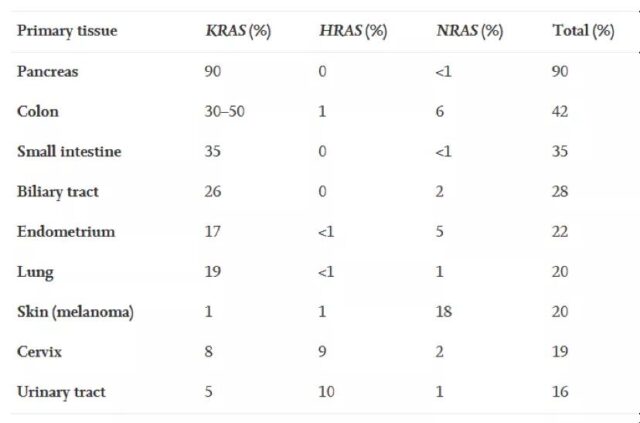
data source: Liuetal.,(2019).TargetingtheuntargetableKRASincancertherapy.ActaPharmaceuticaSinicaB, doi.org/10.1016/j.apsb.
Among KRAS gene mutations, 97% are mutations in the 12th or 13th amino acid residue. The most important ones are the three mutations G12D, G12V, and G13D. Structural studies have shown that most of these gene mutations interfere with the ability of KRAS to hydrolyze GTP.
Three major mutations in the KRAS gene:
- G12D: from GGT to GAT
- G12V: from GGT to GTT
- G13D: from GGT to GAC
The binding force of KRAS to GDP or GTP is very strong, and the affinity coefficient of KRAS to GTP is in the PicoMolar (picomolar concentration) level. When KRAS binds to GDP, there is no kinase activity; when KRAS binds to GTP, there is kinase activity. Wild-type KRAS binds to GDP or GTP, and is regulated by upstream signals.
When KRAS has mutations such as G12D, G12V, and G13D, it will destroy GAP activity. KRAS will continue to bind to GTP, lock KRAS in an active state with tyrosine kinases, and continuously activate downstream signaling pathways (such as PI3K, RAF-MEK-ERK (MAPK), RAL-GEF, etc.). After these downstream signaling pathways are opened, they will stimulate cell proliferation and migration, and ultimately contribute to tumorigenesis.
Let us use an analogy to illustrate the consequences of this mutation: compare KRAS to the custodian of a bank vault (cell proliferation and migration capacity). Under normal circumstances, only the bank president (upstream signal protein) brings the key (activation signal), and the custodian (KRAS) can open the vault (cell proliferation, migration). Now the vault custodian (KRAS) has stolen the vault key and opened the vault door wide, so the vault (cell proliferation and migration) will get out of control.
The relationship between the function of KRAS and tumor treatment
RAS plays a pivotal role in the signal regulation of cell growth. The upstream cell surface receptors such as EGFR (ErbB1), HER2 (ErbB2), ErbB3, ErbB4, etc. [1], after receiving external signals [2], will pass the RAS protein [3] to the downstream [4]. In turn, it stimulates cell proliferation and migration.
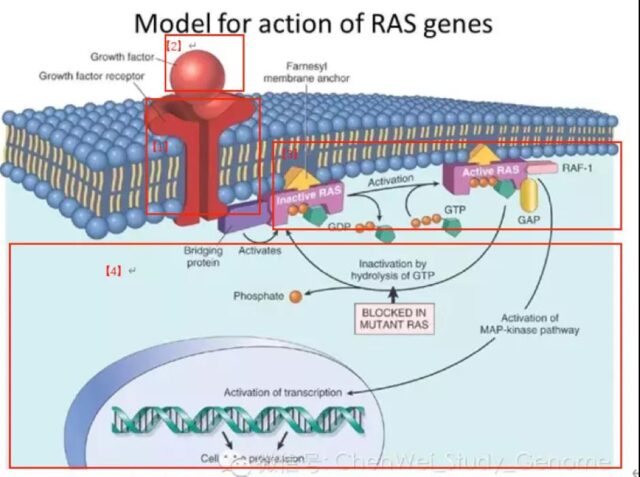
Source: Chen Weixue Gene
KRAS is an important member of the RAS protein. Mutations in the KRAS gene will directly affect the efficacy of anti-tumor drugs targeting the EGFR gene. At the same time, whether there are mutations in the KRAS gene is also an important indicator of tumor prognosis.
If there is an activating mutation of KRAS in the tumor of a tumor patient, then targeted therapies for EGFR, such as cetuximab, Iressa, etc., are usually not very effective. Because KRAS bypasses the inhibitory effect of drugs on EGFR. In addition, tumor patients with KRAS activating mutations generally have a poor prognosis, and the survival period is often significantly shorter than those without KRAS activating mutations.
Therefore, whether there are activating mutations in the KRAS gene in tumors is very important for predicting whether EGFR inhibitor drugs (treatments) will work. Because of this, the detection of activating mutations in the KRAS gene is now one of the important companion diagnostics for tumor-targeted drugs.
Studies have shown that carcinogenic KRAS gene mutations can not only directly promote the proliferation and survival of tumor cells, but also have an impact on the tumor microenvironment. Carcinogenic KRAS induces and affects the cells surrounding the stroma (such as fibroblasts, innate and adaptive immune cells) in a paracrine manner, which in turn promotes the malignancy of cancer.
Tumor cells with mutations in the KRAS gene can secrete a variety of cytokines, chemokines, and growth factors, including IL-6, IL-8, IL-23, CCL9, hedgehog and so on. These factors can reprogram stroma cells in the tumor microenvironment. For example, IL-6 and IL-8 can maintain the inflammatory phenotype of the stroma in pancreatic cancer and lung cancer. Granulocyte-macrophage colony stimulating factor (GM-CSF) secreted by tumor cells can stimulate myeloid-derived suppressor cells (MDSC) to infiltrate the tumor, thereby inhibiting the anti-tumor immune response.
The effect of KRAS mutation on tumor microenvironment
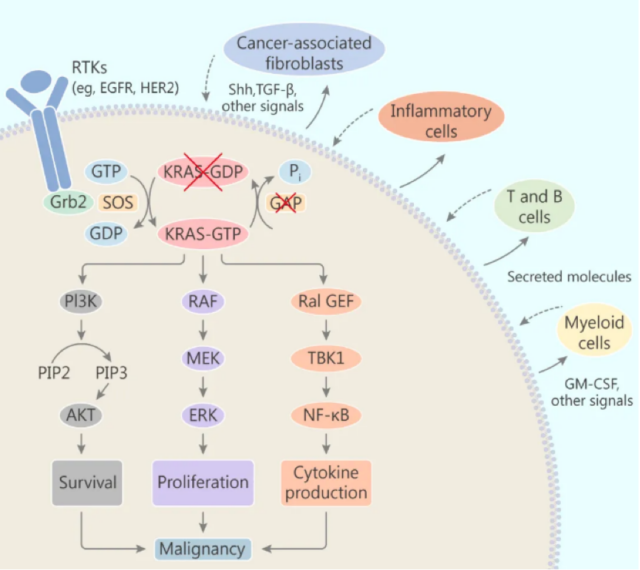
Data Sources: Liuetal.,(2019).TargetingtheuntargetableKRASincancertherapy.ActaPharmaceuticaSinicaB, doi.org/10.1016/j.apsb.
Genetic testing for KRAS mutations
In January 2012, the FDA approved Qiagen’s KRAS mutation detection method as a companion diagnosis for cetuximab. In May 2015, the FDA approved Roche’s KRAS mutation detection method for the diagnosis of metastatic colon cancer.
China’s Xiamen Aide Company, Wuhan Youzhiyou Company, Wuhan Haijili Company, Suzhou Weizhen Company, etc. all provide detection kits for KRAS gene activating mutations.
The above-mentioned detection methods for KRAS mutations are based on the principle of real-time fluorescent quantitative PCR method.
KRAS, a non-medicable target?
Since KRAS is so important and the cancer-causing mutations in the KRAS gene are very clear, why hasn’t a targeted drug that directly targeted the KRAS gene been marketed so far? The reason is that the KRAS protein is a featureless, nearly spherical structure without obvious binding sites, and it is difficult to synthesize a compound that can target binding and inhibit its activity. Unable to conquer for a long time, KRAS has become synonymous with the “unable to drug” target in the field of oncology drug research and development.
The difficulty:
1) KRAS has a wide range of effects. The normal activity of KRAS is also an activity required by many normal cell functions. If a drug that directly inhibits KRAS is selected, the drug may be very toxic and its side effects may be strong. Moreover, KRAS has high homology with NRAS and HRAS, and drugs that can inhibit the activity of KRAS are likely to inhibit the activity of NRAS and HRAS. Then, the toxicity of this drug may be great.
2) The currently known active functional domains of KRAS are mainly pocket-like functional domains that combine KRAS with GDP or GTP. Different from the weak affinity between protein kinase and ATP, KRAS binds to GTP or GDP very strongly, and the affinity coefficient reaches PicoMolar (picomolar concentration, 10^-12) level. The concentration of GDP and GTP in normal cells has reached the MicroMolar (micromolar concentration, 10^-6) level. Therefore, the normal concentration of GDP and GTP in the cell is 10 times higher than the concentration required for KRAS binding. However, RAS lacks a large enough pocket that can bind small molecules; therefore, it is very difficult to make a small molecule compound whose binding ability with KRAS can match GDP or GTP.
3) Designing an active drug that only specifically inhibits the mutant KRAS protein, and minimizes the influence on the activity of the normal KRAS protein. This compound is required to have a good selectivity for the mutant KRAS. This is another problem in drug design.
4) However, strategies for indirect targeting of KRAS are also difficult, including blocking KRAS cell membrane positioning and targeting signal molecules downstream of KRAS, such as family members such as RAF, MEK, ERK and PI3K. Specifically, the difficulties of indirect pathways include: (1) RAS is an essential pathway related to normal cell growth and survival. Targeting the essential pathway first faces serious toxic and side effects, resulting in a very narrow or no therapeutic effect index; (2) compensation escape mechanism , And (3) Signal feedback and redundancy due to strict regulation.
In summary, it is the main reason why there is no targeted drug directly targeting KRAS on the market so far.
Strategy and progress of drug development targeting KRAS
1) Directly targeting KRAS mutants-breakthroughs in recent years
In recent years, breakthroughs in the study of covalent inhibitors for KRAS mutants have made it possible to target KRAS mutants through allosteric sites. In the KRASG12C mutant, small molecules covalently bound to the mutant cystine are more likely to bind to the KRAS protein bound to GDP. This combination reduces the affinity of GTP and KRAS, and at the same time prevents GEF from catalyzing GTP to replace GDP, locking the KRASG12C mutant in an inactive state. The discovery of this binding “pocket” on the KRASG12C mutant has spawned a variety of small molecule covalent inhibitors that target the KRASG12C mutant.
Among them, Amgen’s AMG510 is the first KRASG12C inhibitor to announce clinical trial results. MiratiTherapeutics’ MRTX849 has also entered phase 1/1b clinical trials. The company’s other KRASG12C inhibitor MRTX1257 is still in the preclinical development stage. The IND application for ARS-3248 developed by Wellspring Biosciences has been approved by the FDA. JanssenBiotech will take over the phase 1 clinical trial and subsequent clinical development.
In addition to these KRASG12C inhibitors that have entered clinical development, Boehringer Ingelheim recently published a study on PNAS that the company has discovered a small molecule inhibitor that can bind to the KRAS protein at the nanomolar level. The “pocket” combined with the small molecule inhibitor of BI-2852 is different from the existing KRASG12C inhibitor. Its binding to KRAS can affect the binding of KRAS to GEF, GAP and downstream effectors. In in vitro tests, this inhibitor can play an anti-proliferative effect in cells carrying KRAS gene mutations. Boehringer Ingelheim plans to develop a more potent and specific KRAS inhibitor based on this inhibitor.
Mirati also has a KRAS inhibitor designed to inhibit KRASG12D mutants.
2) Indirect-a protease that regulates the binding of RAS to the plasma membrane
Such as inhibiting the activity of farnes acyltransferase and PDEδ
KRAS is located on the cell membrane through a farnesyl group. The KRAS protein can only be better activated by the upstream signal protein when it is located on the cell membrane. Therefore, scientists consider whether to inhibit the farnes acylation of KRAS protein by inhibiting the activity of farnes acyltransferase, thereby reducing its localization To the cell membrane, achieve the purpose of inhibiting KRAS activity.
At present, some compounds targeting farnesyltransferase have been designed, but these compounds have poor tumor inhibition effects in later experiments.
3) Indirect-target proteins that regulate KRAS activity
Due to the “non-druggable” nature of KRAS protein, targeting proteins that regulate KRAS activity has become a research direction for indirect targeting of KRAS. GEF can catalyze the binding of GTP to KRAS, so inhibitors that inhibit the function of GEF can indirectly reduce the activity of KRAS. Currently, Boehringer Ingelheim has a SOS1 inhibitor in its R&D pipeline. SOS1 is a GEF that catalyzes the combination of GTP and KRAS.
Another target of interest is SHP2, which is a protein tyrosine phosphatase that regulates the RAS/MAPK signaling pathway. Scientific research has shown that inhibition of SHP2 activity can be combined with other treatments including MEK inhibitors to enhance the killing effect on tumor cells carrying KRAS gene mutations. Novartis and RevolutionMedicines both have SHP2 inhibitors in clinical development. Novartis has also recently reached a cooperation agreement with Mirati to test the effect of the company’s SHP2 inhibitor TNO155 in combination with Mirati’s KRASG12C inhibitor MRTX849.
4) Indirect inhibition of KRAS
Another way to bypass the lack of small molecule drug binding targets in KRAS protein is to indirectly KRAS protein through innovative treatment models. In this regard, targeted protein degradation agents, RNAi therapy, antisense oligonucleotide therapy and cancer vaccines are all directions that researchers are exploring. Currently, protein degradation therapy and RNAi therapy targeting KRAS are still in the early stages of development.
The antisense oligonucleotide therapy AZD4785 targeting KRAS developed by AstraZeneca and Ionis has been discontinued. In terms of cancer vaccines that target KRAS, Elicio’s KRAS cancer vaccine ELI-002 can trigger a strong immune response to KRAS protein mutants in preclinical trials, causing T cells to produce powerful cells against target cells expressing KRAS mutants Cracking function. Moderna also has an mRNA cancer vaccine targeting KRAS in Phase 1 clinical studies.
5) Indirect-inhibition of RAS effector signaling (eg, RAF and PI3K)
In view of the difficulty of designing inhibitors that directly target KRAS, many scientists put more energy into indirect methods such as designing targeted drugs for the signaling pathways downstream of KRAS. For example, design targeted drugs for B-RAF. Targeting RAS downstream effect signaling
Blocking downstream effector signaling is one of the most attractive strategies for targeting RAS. However, for at least 11 different downstream effector signaling pathways, which effector to target and whether multiple effectors need to be inhibited at the same time are still critical issues.
The two effect pathways that have attracted the most attention are the RAF-MEK-ERK mitogen kinase (MAPK) signaling pathway and the PI3K-AKT-mTOR pathway. It is known that mutations in genes encoding components of these two pathways (BRAF and PIK3CA) are important drivers of carcinogenesis, and they are also targetable protein kinases. However, BRAF and PIK2CA mutations do not overlap with RAS mutations in many cases.
They are relatively independent downstream driving carcinogens. Targeting BRAF or PIK2CA by screening biomarkers also does not refer to the mutation status of RAS. Therefore, strictly speaking, targeting BRAF and PIK2CA is not the main method for targeting RAS mutations.
The first-generation RAF inhibitors (vemurafenib and dabrafenib) have been approved for the treatment of BRAF mutant melanoma. However, when these inhibitors were evaluated in RAS mutant cancer cells, abnormal activation of ERK signaling was observed rather than inhibition. Later, it was found that the first generation of RAF inhibitors only inhibited BRAF, not only did not inhibit CRAF, but caused the activity of CRAF to increase, thereby activating the MEK-ERK signaling pathway. The new generation of RAF inhibitors include LY3009120 and PLX8394, which are broad-spectrum RAF inhibitors, and clinical trials are ongoing.
In addition, there are indirect targeted RAS synthetic lethal strategies, and interference with the metabolic process of cancer cells regulated by RAS, which are mostly limited to the proof-of-concept stage, which is far from a drug.
(source:internet, reference only)
Disclaimer of medicaltrend.org



